What's New in MacVector 15.5?
Overview
MacVector 15.5 introduces an entirely new way to view the results of BLAST searches, with an interactive graphical map allowing you to view the context around each hit and download matching segments. Unannotated Open Reading Frames are now automatically displayed whenever you open a DNA sequence. A number of changes to the Align To Reference and Contig Editors simplify the task of resolving repeat sequences during NGS genome assemblies. A variety of menu changes help make some of MacVector's features more accessible to less experienced users. A number of optimizations have been made to better handle opening and analyzing extremely large sequences (up to 1,000 Mbp or more) and alignments You can view the release notes here.
BLAST Map Results:
There is a new graphical BLAST results tab. It displays a graphical representation of the query sequence aligned against each high scoring segment pair of the BLAST hits (NOTE: individual hits may have multiple high scoring segment pairs). This allows you to clearly see which genes or features match your query. If there are too many features to display in the graphical pane, simply hovering over it will expand to display all of the overlapping features. For each hit, you can choose to download the entire sequence, or, particularly useful for hits to entire genomes, just the aligned segment plus ~2kb either side.
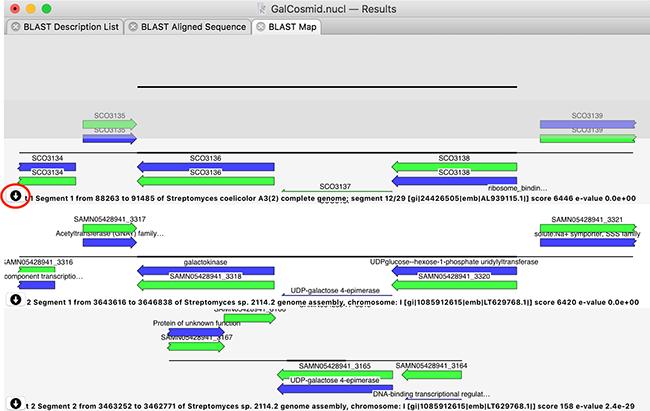
This interface represents a new and powerful approach to quickly identifying the function of your query sequence. With the advent of NGS genomic sequencing, bacterial query sequences in particular typically match entire genomes – this allows you to identify and download just the region that matches your sample sequence for further detailed analysis within MacVector.
Automatic ORF Display
MacVector now automatically searches for open reading frames in every DNA sequence that you open. This works similarly to the existing automatic Restriction Enzyme display. There is a new DNA Map tab in the main Preferences window that controls both of these functions;
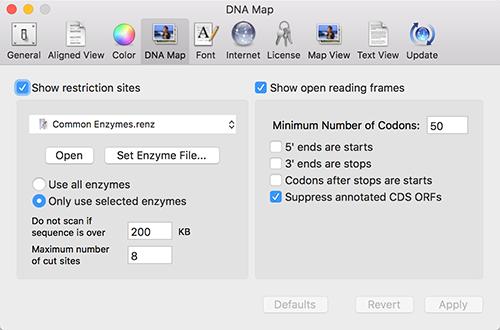
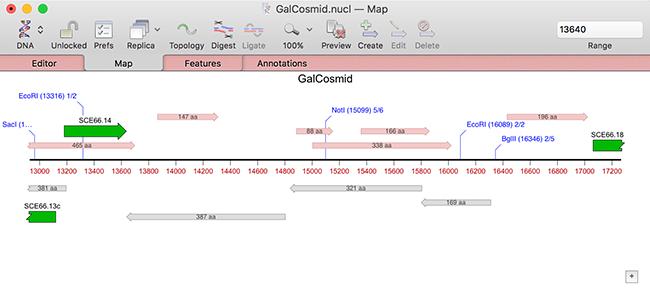
Note that there is a checkbox (turned on by default) to suppress the display of ORFs that are already annotated on the sequence. In addition, the default appearance of ORFs has been changed to be less obtrusive in crowded Maps and they are now labeled with the length of the ORF;
Align To Reference Editor Context Menu
Both the Align To Reference Editor and Contig Editor now have context sensitive menus. To invoke these, simply right-click (or <control>-click) in the window. Many of these functions have been specifically designed to simplify the use of MacVector for closing the gaps in bacterial genome sequencing projects. The functions available for the Align To Reference Editor are;
Export Consensus with/without Gaps
Align Selected Reads
Delete Selected Reads
Reset (unalign) Selected Reads
Export Selected Reads as FASTA/FASTQ
Select Matching Pairs (*)
Extend Reference with Selected Read (**)
(*) – if you have aligned a set of paired-end reads, you can select individual read(s) and use this function to select the corresponding mate(s). This is particularly useful if you want to find pairs that will extend a contig and export them for further analysis/assembly.
(**) This is active if you have selected a single read that hangs over either end of a Reference sequence. This will extend the Reference in the appropriate direction using the sequence of the read.
The Contig Editor has a subset of these functions with just the Export... and Select Matching Pairs functions available.
Miscellaneous Changes
Map windows now update when you change the default symbols for features.
A number of menu items have been renamed to more clearly explain what they do.
ORF results can now be filtered by minimum length.
Some bugs saving large assemblies have been fixed.
A number of issues selecting different display options in the Phylogenetic Reconstruction tree display have been resolved.
A few intermittent crashes when editing Multiple Sequence Alignment documents have been fixed.
MacVector now does a better job of interpreting complicated Bowtie alignments so that the alignment display has fewer out-of phase mismatches, whilst still retaining all of the residues in each read.
There is now an option to control the length of vertically oriented labels in the Map tab.
A bug where you could not display multiple digest when just a small number of restriction enzymes were used has been resolved.
By default, only the main three NCBI databases are now shown in Entrez searches. You can ask to view more, but you may not be able to retrieve sequences from many of the others, other than through a browser.
A number of default settings have been changed to enhance the interface experience for new users.
|

