
Update 19 August 2013: We’ve added support for Muscle and T-Coffee to the MSA editor
We get a lot of comments and questions from users on the various alignment functions in MacVector. They say there’s more than one way to skin a cat (not that I’ve done that – I have skinned a catfish, but I only know one way), and thats certainly true for alignments in MacVector. Each function is designed for a different purpose. First, lets just list the functions;
- ClustalW – we also call this the “standard” Multiple Sequence Alignment (MSA)
- Align to Reference
- Pustell Matrix (also known as a Dot Plot)
- Internet BLAST
- Align to Folder
- Contig Assembly
ClustalW/Multiple Sequence Alignment (MSA)

If you have two or more related sequences (DNA or Protein) and you want to examine the relationship between them, use this function. Choose File->New->Protein Alignment (or File->New->Nucleic Acid Alignment) to create an empty MSA window. Add sequences to the alignment by using the Edit->Add Sequences from File menu item then click on the Align toolbar button to automatically align the sequences using ClustalW. Click on the Prefs toolbar button to control the appearance and behavior of the data in each of tabs that represent different views or analyses of the alignment. This functionality is most suited for protein alignments, or for nucleic acid sequences where you are interested in examining phylogenetic relationships. If you wish to compare two or more DNA sequences, you should definitely consider if one of the other alignment functions may be more suitable.
Align to Reference
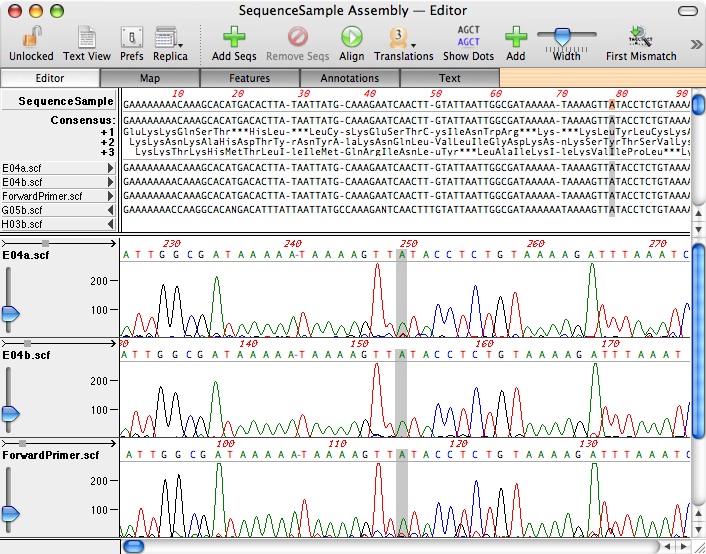
Use this if you have a reference sequence and you want to align one or more DNA sequences against it. A typical use would be in resequencing e.g. sequencing a cloned PCR fragment to check no errors were introduced, sequencing across end junctions, scanning for successful mutagenesis clones etc. In each case, open the file that represents the parent or “reference” sequence, then choose Analyze->Align to Reference. In the window that opens, click on the “+” button to add sequences from disk – these can be in any format that MacVector can read – typically ABI or SCF chromatogram files, but you can add plain sequences as well. When you click on the Align button, choose the Sequence Confirmation algorithm – this is tuned to expect the small insertions/deletions you would expect in raw chromatogram files. Compared to ClustalW, Align to Reference has the advantage that it will automatically “flip” sequences to guarantee optimal alignment.
Align to Reference can also be used to align cDNA clones against a genome sequence. The steps are similar – use the genomic sequence as the reference, then add one or more cDNA clones to the alignment. Again, these can be chromatogram files. Now choose the cDNA Alignment algorithm when you Align – this is tuned to expect large insertions representing the intron regions.
Pustell Matrix
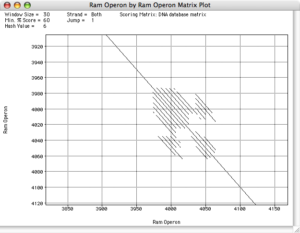
This “Dot Plot” function is great for identifying weak regions of similarity between two sequences. It is not designed to show full-length alignments between two sequences, but instead shows shorter segments of direct or inverted similarity. You can use this to identify shorter regions of similarity, then copy those sections to new sequence windows for more in depth analysis using ClustalW or Align to Reference. Dot Plots are also the best way of identifying sequence rearrangements – the display clearly shows insertions and deletions (the main diagonal will be broken and have an offset) and even inversions (the inverted diagonal will run bottom left to top right and be colored blue). Finally you can use it to identify repetitive regions which appear as parallel diagonals offset from the main diagonal. Pustell Matrix can be used not only to compare DNA:DNA and Protein:Protein, but you can also use it to compare DNA:Protein where the algorithm will translate the DNA in all 6 frames before aligning to the protein.
Internet BLAST
Use this to identify and align a test sequence to the databases at the NCBI using the popular BLAST algorithm. One slightly hidden function in MacVector is that you can select sequences in the “hitlist” and then choose Database->Retrieve to Disk or Database->Retrieve to Desktop to download the matching sequences from the NCBI. You don’t even need to select the entire line – just select part of a line and use the Retrieve menu item.
Align to Folder
This allows you to scan a local folder full of sequences (in any format MacVector can recognize) and align them using the FastA alignment algorithm. Its kind of like a local BLAST, but more sensitive. Like the Pustell Matrix, you can choose to search DNA with Protein and vice versa. Many users like this function because the text alignment output also shows the features in the test sequence. This can be very useful for demonstrating the differences between your sequence and other sequences for patent purposes.
Contig Assembly
This requires our optional Assembler add-on. Use this if you want to align two or more DNA sequences with the idea of assembling them into a longer sequence with a consensus. Its is primarily designed for de novo sequencing, where you have no reference or scaffold sequence to align the individual sequences to. The MacVector implementation uses the popular phred, phrap and cross_match algorithms from the University of Washington that use quality values for improved accuracy of assembly. While you can use this for resequencing, you should consider whether the Align to Reference function might be a better choice.
Tutorials
There are tutorials for Sequence Confirmation and Contig Assembly in the Documentation folder of your MacVector installation. You can also download copies from our website.
So there we have at least five ways to align sequences using MacVector. Now if I can just find another 4 ways of skinning a catfish (or even just ONE thats easier than my current method) then I’ll be all set.
Comments
One response to “Alignments in MacVector”
Sry for writing OFFTOPIC – which WP template are you using? It looks interesting!