
MacVector’s Import Features tool allows you to import annotation from many Genome Browsers (e.g. Ensemble, UCSC, etc). MacVector can annotate an empty or annotated sequence.
BED, GFF, GTF, and GFF3 formats
GFF, GTF, GFF3 & BED files are all file formats that are used to store annotation (features) generally without containing any sequence. Although it is common that they will be accompanied by a fasta file containing the sequence only. They emerged as a way of exporting, or exchanging, information from a specified region of an entire genome without having to take the entire genome.
Most sequence formats were developed to be for a specific gene or protein. Although this is no longer true they are still orientated to be of a region of fixed length. These annotation files are not at all length specific and could potentially store just two features that were at either end of the same chromosome. They are a much more flexible way of dealing with annotation, especially a large amount, than a fixed length sequence format such as Genbank.
They also are not limited to a single sequence and can contain information from multiple sequences in the same file (Fasta files can also contain multiple sequences). For example you could store the entire human set of chromosomes in a pair of (quite large!) files. A multiple sequence Fasta file and a single GFF file.
The format of these annotation files does vary (who ever said Bioinformaticians had to be consistent!) but basically their format consists of a set of individual lines (one line per feature) along the following lines:
SEQUENCE ID, START, STOP, FEATURE TYPE, NOTE
Sequence ID is the sequence these annotations belong to.
START and STOP are the region of sequence they are annotated against
FEATURE TYPE is obvious! Note that this does not always correspond to a correct Genbank Feature Keyword
Genome Browsers
These tools (generally online web gateways) allow you to browse the entire chromosome or genome of a particular organism. Almost like a graphical model of a sequence database. All the information known about that particular organism’s sequence that has been submitted to one of the large sequence databases (e.g. Genbank at the NCBI) should be visualised within the genome browser. You can download all the annotation contained within a particular region fairly easily using one of these annotation formats. Then you can either annotate an existing file that you are working with (so preserving your own “private” annotation with all known public annotation).
To annotate a sequence with a BED/GFF/GFF3/GFT file in MacVector
From the UCSC’s Genome browser
- Click on this link to open the UCSC’s Genome Browser.
- Select C.elegans (or click the link to “worm”) and enter sel-12 in the gene name. Click SUBMIT.

The interface will change and show all annotation associated with that region. You can modify the amount or type of annotation being showed. This particular gene, C.elegans Sel-12 is located on Chromosome X
- Click the Tools then Table Browser menu link at the top of the page
This will now allow you to export all the annotation associated with the previous displayed region (tracks).
- Change the REGION to POSITION.
If it is left at genome the entire genome will be downloaded
Change the OUTPUT FORMAT to GTF or BED
Click GET OUTPUT
Now switch back to MacVector
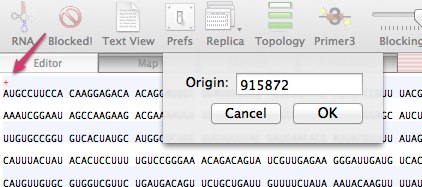
Now you need to open the sequence you want to annotate. For this example we could go to DATABASE > ENTREZ and search for and download Accession Number U35660. However, that only contains the mRNA and not the genomic sequence. So instead download the fasta sequence from the NCBISequence : Chromosome: X; NC_003284.7 (915873..918235)You will need to ensure that the start position of the downloaded file corresponds to the start of the region of the chromosome we have just downloaded the annotation for. This is easily done in MacVector.
Double click on the RED cross located near the start of the sequence in the Editor View and change it to 915872
Now we will import our downloaded annotation.
Select FILE | IMPORT FEATURES
The Sequence ID (SeqID) contained within all features in the file will be shown in a dialog along with the number of features for each SeqID and the region of the sequence that these will be annotated against. A warning will be displayed if any of the features are outside of the region to be annotated.
Select the appropriate SeqID ChrX and click OK
A dialogue will be shown with the number of annotations that have been added. If you annotate a blank sequence (e.g. a fasta file) the resulting features may be initially hidden. However, you can easily show them from the Graphics Palette tree view.You can choose to annotate your sequence with all the features contained within the imported file or to ignore duplicates.
Click OK
ToxoDB Genome browser
Here’s a similar workflow from the ToxoDB Genome browser.
For this workflow we’ll start with a “random” 2kbp sequence from TaxoDB as a “starter” empty sequence.Toxoplasma gondii ME49
"CCTTCCCTGCGTCAGAGGAGAAGAGAACGGCTTGACCGATGGAGGACCCCGCAAACATGAGGGCGAAGGTAGTCTGCATGATCTCTGAACAAGGAACACGGCGCGGAAAGGGAAGCACAGAAGGAAGTCGATCAAGACACCCTGCGTTGTTTTTCGGGGAGCCCCAGAGAGGGAGCTCGCGGCTCTGGACTTCAAGGTCCGTCGAGCAGCAGAACGCTTCACTCGGCAAGGAAGGAGCAGTTTCTTCTCTCGCGTCTTGTTTCGCTTTCACGGCTTCGTTTTCTCGCCGCGACCTGCGAAAAGAAAACAGCTCCCCTATAAGAACTCGACTCTCGAGCCTGCGGTTTGGTATCGGCTTTTTCTTCAGAGTTTTTTCTGTCGCGCGTTCGGACAACCAGTTCCGTGCTTGCGCGCCTCCTCTGAAGGCCGCGCCCGCCTCTCGACTCCCGTCGCTTCTCTCTTCGGCTGGATAAGAGAAAACGCTGAACGAAGAGGAGAGTACGCACTGGCATCGTTTGTCGACTTTCGTCTCCAGGTGGGGGAGTGTCGGTCGACTCACCCAAGGGATTCTTCCCTTCGCTGCTCACGATCTGGCCGCCATACCAAAAAATCAGCGCCTGCAGAGCGTACTGAGCTCCCTGACAGACACGCAGACGCAGCGGCAAGGAGACGCTGAAAAGAAGAAAGACAACCGGAGAGCGCGGAGAACAAAGAACTGTGAGCGTGCAACGACGGGATCAAGGACGACAGCGAATCTCCCGTCTTCAGGACCTCGACGGGCATTCCGCATGGCAGGTCCTTTGACTCCGAAAAACTCTGCGGCAGCCTCGATGACCCTTACCCCCCCGGAACATCCCCGAGAGCTCGGTGGAAAAAACCTCTCATTCGAGAGCGACAGATCAGGCTTTGCTAGTCGAGCCAGAAGGCAGGAGAAGGAACGGAGCGAACCGCGGATGCGTCTCTCTGCGCGGACGAGTCTTCATGAGCAGGCACCGCGACGTTCCAAGAAGCAGAAAGAGAGAGAGGAGAGAGGAGAGAAGCGAGAAGCTCGGGAACTCACGAGGAAGCAACAAAGATCTCTCCTCGTCACTCACGTTCCGTCGACCTGCATGGCAGGCGTGACGCGGCATGCACAGCAGAAGACCTTTCGAGGTCACCACACACACCGCCTCGGACGTCGAGAAGTCTCGATCTTTGTGAACCACAGGGCTCTGTTTTGTGTGGCGGAACGAAGAAACCAAGCGCTTAGGATGGAGCTCACTGGAGAGCAGGAAACGGATCTTCAAACGAGTGTCGACGTCCTCCCGCGCATCCGAACCGAAACTCAAACGCGCTCCAGAGAGACGACATAGAAGACAGAGACGTACAATGAGAGAAGAAGAGACAACGCGGCAGGGGGAGTCTGACGTCCGACCTCGACTCGAGAAGTCGCTCGCCAAAACGTGTGTGCAGTGTCTTCTGTTTCTTTCCAAGTTCTCCAGTCCGAAGAAACCGGACACTCTGACATGACTCGATACAGGGACCTGCCCGCCGACTCTTTCCTACTTTCAGCGGTCCTCCCTGTTCATCTTTCCTGTGACATTTCGGCATCTCTTTTTCTTGGTTTCCTCGCCTTCTCACCTGACTGAAGCCCCAGAAAAAGCCGAGGAGAGCCGCAGCGCGCTCTTCTTCCTTCAGCGTCCTCAGAAGAACGCTCTGGTACCGTTCCGTGAAGTGAGGCTCTAAACCGAACGCTGAAACAATGCGAATACCGTTCAGAGCCTCGCTCATCACGAAGGCAGCGGTGTCGCGGTCCTCCACCTTCTCCGCCTTCTTGTTCGCCCCCTCACCTGCAGAAAAAACTCCAAGGTTTCCAAAGCCTCGTGAGGCTCCCCATGAAGCTCTCCGCCTACGCTTGCGCAGATTGAGGCAGAGCAAACTACGCAAATGTGAGCCTACATGTACACACAGTTTCGTCGAGATTTGTACCTATATCTAAGAAGATTTGTACGGAAATGCGGGTGTGAAGCGGCAGTTTTCGAGGTGGCGTGCATACATCGACGCGACTCGGAGACCCAGCTTTGAGGAGACAGGAGAGAGAAAAGGAAACGGAGATAGAGCAGGTGGGGAGATCAGGTTTGCTCTGGGAGACGTGGACGGTCGCAGACGAAGAAGCAGACGCACGGAGCGAGCAGTGCAAAAAAGCGCGAGACAGAGCCGGCGCTGGGGAACTTCTGAGGAAGAATTCGAGAGAGAGAGGACCGTGGTGAAAAGCCAAGCTAAACGCGTGGACTTCCAGTTCTGCGGACTTTTCGGAGCCGAAATGTGAGACTGAGCGGCAGTGGCGGGGAGCAGAAGAACAAAACATGCGAGGACCACGGCGGCCAAGCGCGCGTCTCCAAAGAACGCGATGATGACACCTGAAATAAAGATAATGGAAAAAACAGAAGAAAAGCAACGGTCTCTCGCGCAGTCTTCAATCACGAGTTGACGCACACACGCATTAGGGGAGAACAACCTCTGCGATGTTGGATGCTTCTTTAGTGAGTGGACGTTTCCAGAAATCAAATCAAAGTAGCACGACACCGACAAGCAAAGAGATGTATACTTTCGTTCACGAGCACTGAAAGACGCGTCTAGACGCCTACGGATGCAGAGGGATCTGAAGCGACGAGTGAACAGTCAAAAAGCTTTCCGGCCTACCAGTGACAACAGCAGCCAATCCCTGGGTCATCGCGAGTGCGTTTCCAGCGCTTCCTGTCTTGACGAGAAGGACGTCGCTGGAGAGAACTCCCGTGAGATATCCTGAGGTCATTTTTTAGAAGAAGCGAACACTCTGGCGCGGCGTCTTCACTCTCGTGCTCACAGAAAGAATGAACTCACGAGACCCATGGCAGGACAAGTCTCAGACAGACACACAC"
Blast the above sequence using the Blast interface at Toxodb.org.
This will find a single hit and display a link
Click on the link to open it in the ToxoDB Genome Browser.
Select DOWNLOAD TRACKS, then CONFIGURE and change it to GFF3 format and SAVE TO DISK. Now click GO.This downloads a file “dumped_region” which will contain all the annotation stored in the ToxoDB Genome Browser in GFF3 format.
Now switch back to MacVector and open the 2Kbp sequence.
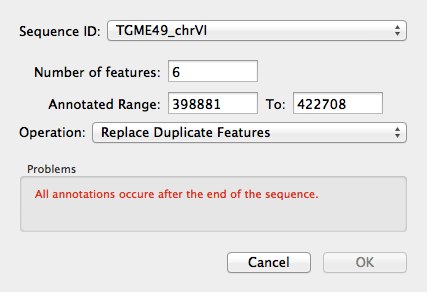
You can use FILE > NEW FROM CLIPBOARD to bring the sequence into MacVector quicklyAgain if you do not change the start coordinate of the sequence the IMPORT FEATURES dialogue will show an error
Change the start coordinate of the sequence to match its location in the genome (which is 405235 as detailed on the Blast hit page) as in the step in the previous workflow.
I then went to FILE > IMPORT FEATURES and selected the GFF3 file.


Keeping a sequence updated
You will be able to “update” and add new annotation to your existing sequence. For example after a few months I could revisit these two genome browsers website and download an updated GFF3 file. Upon importing these features it will optionally replace any duplicate features and add new ones. So you can work with a sequence and also keep it updated as other researchers find more about this particular sequence.
Duplicate Features
Due to the lack of strict standards across the many different file formats it may be that a potential duplicate is not recognised as such because the wording or keyword is different. In the majority of cases some degree of manual curation of the annotated sequence will be required. In all cases MacVector will err on the side of caution and will never throw away any potentially interesting or important information contained within a feature. Only entries that are 100% the same (after being parsed during the import) will be considered as duplicates. MacVector will never class a feature as a duplicate if the START, STOP or FEATURE TYPE are different in any way. Even if they differ by just a single base.
Comments
2 responses to “Importing features from a Genome Browser”
I am having difficulty uploading a custom UCSC genomce browser track imported into MacVector 10.6. Can MacV accomodate tracks that consist of ChIP quantitative data, or will it only represent a flat feature?
Hi Mark, Currently MacVector will only support flat features as defined by the NCBI, DDBJ and EMBL:
The DDBJ/EMBL/GenBank Feature Table Definition
We do this for compatibility reasons and to avoid “data lockout”. A MacVector file can always be exported to a 100% compatible Genbank file. However, I will add this to our enhancement request database.