
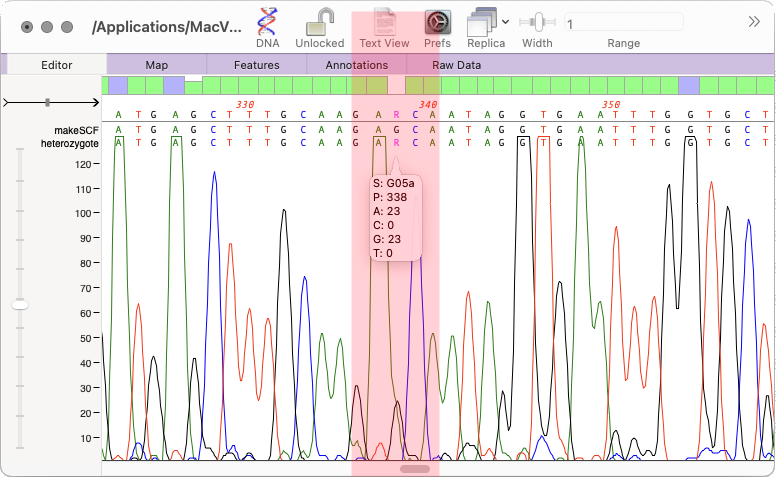
In MacVector 18.5 we added a new tool to call heterozygotes in sequencing reads.
The heterozygote analysis tool allows you to either view heterozygotes in Sanger trace files or to permanently change the basecalled sequence with an ambiguity representing the called heterozygote. The tool works on multiple trace files in the Assembly project manager or the Align to Reference editor. You can also run it on a single trace file in the Single Trace Editor

How to run Heterozygote Analysis
To view putative heterozygotes
- Run ANALYZE – HETEROZYGOTE ANALYSIS
- An Options dialog will appear. Change the options and click OK
- A summary dialog will appear showing the number of heterozygotes found across how many sequences.
- Click OK
- A new tab will appear in the Results window showing the location of the possible heterozygotes.
To permanently basecall the sequence
Assembler
- Add trace files to your Assembly Project
- Select one more more trace files
- Select ANALYZE | BASECALL | USING HETEROZYGOTE ANALYSIS
- An Options dialog will appear. Change the options and click OK
- A summary dialog will appear showing the number of heterozygotes found across how many sequences
- Click OK
Align to Reference
- go to FILE | NEW | ALIGN SEQUENCES TO A REFERENCE
- Choose your reference sequence
- Add trace files to your Assembly Project
- Select one more more trace files
- Select the BASECALL toolbar button or ANALYZE | BASECALL | USING HETEROZYGOTE ANALYSIS
- An Options dialog will appear. Change the options and click OK
- A summary dialog will appear showing the number of heterozygotes found across how many sequences
- Click OK
Trace File Editor
- Double click to open a trace file.
- Select ANALYZE | BASECALL | USING HETEROZYGOTE ANALYSIS
- An Options dialog will appear. Change the options and click OK
- A summary dialog will appear showing the number of heterozygotes found.
- Click OK
Settings
- Normalise peak heights using a window of (25) residues
- Percent of each peak width to use (50%)
- Minimum number of normalised residues (3)
- Minimum Heterozygote threshold (35%)
- Minimum Base Call threshold (75%)
- Ignore low quality regions (yes)
- Where a window of (21) residues
- Has an average quality value of less than (20)
You can suppress normalization, which the algorithm uses to reduce false positives by comparing peaks around a region of sequence rather than an individual peak. However, this may miss heterozygotes if the combined peaks are significantly lower than surrounding peaks. Disabling this setting will increase sensitivity such as when a genome has multiple copies of a gene, but a SNP exists in only one of those copies.
