
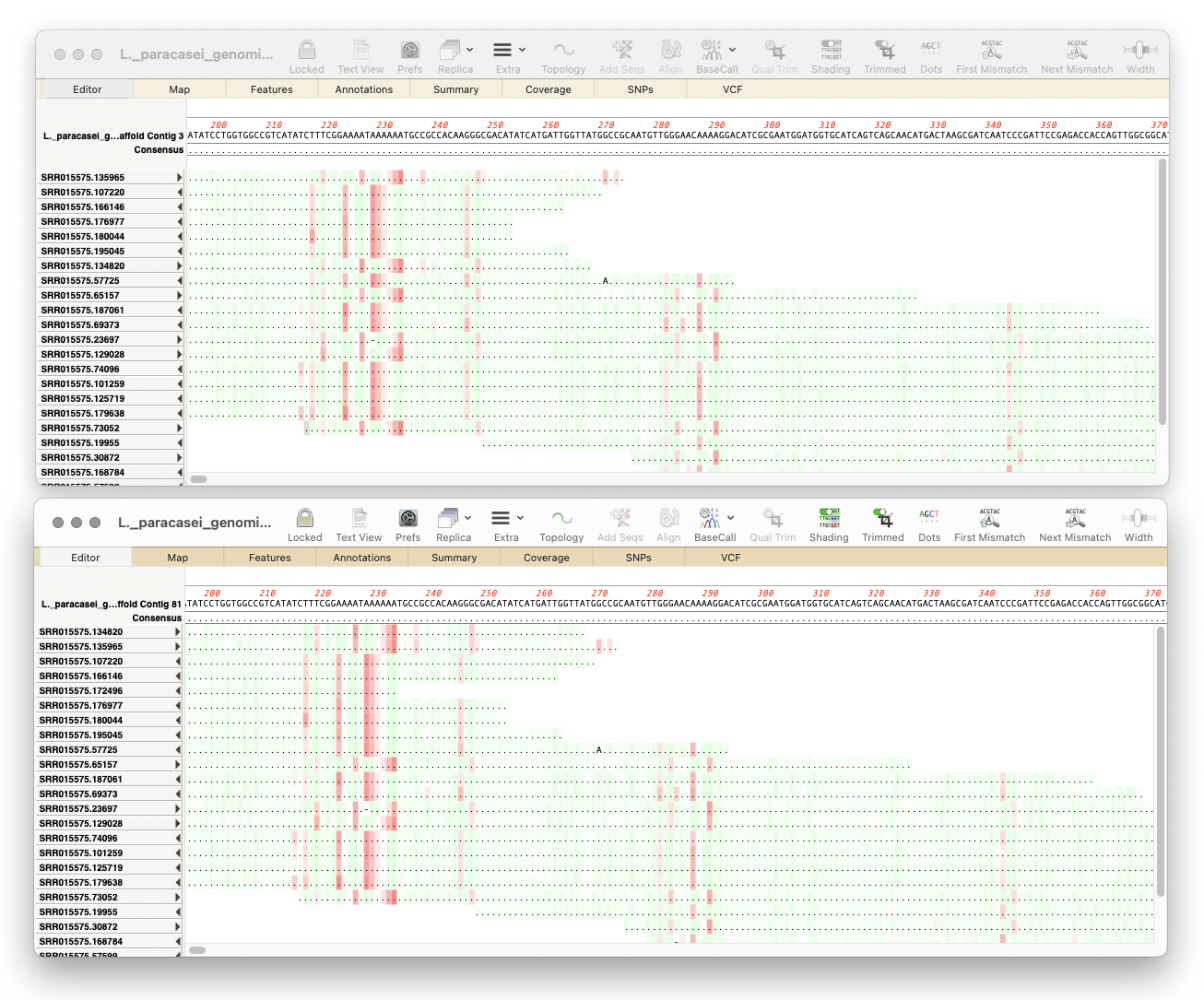
Here’s a few tips regarding analyzing long read data from the Oxford Nanopore Technology MinION and GridION machines.
First, you should always first create a File | New | Assembly Project and then (typically) click on the Add Reads toolbar button and select the appropriate fastq formatted data files. These are often supplied in compressed “.gz” format and can be used in MacVector directly without the need for decompression.
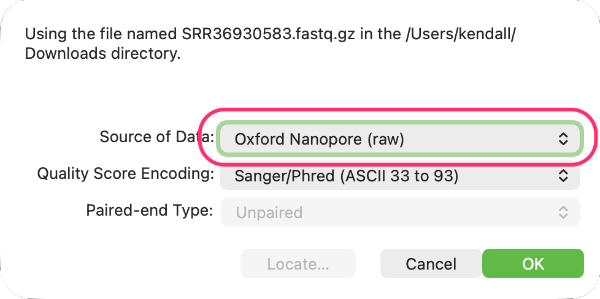
Second it is VERY IMPORTANT to let MacVector know that the fastq files contain Oxford Nanopore data. To do this, double-click on the Status cell (which by default will indicate the files contain Illumina data) and select Oxford Nanopore (raw) from the popup menu.
Now you can run various analyses directly on the imported files.
- de novo Assembly: If you just want to assemble the reads into one or more consensus sequences, choose Flye from the Assemble toolbar button. This assembler is optimized for noisy long read data. There are also options to “polish” the resulting consensus sequences using Flye itself or a separate polishing application called Racon. This can be slow, so be patient, but ~10Mbp assemblies tend to run in 30 minutes or so.
- Reference Assembly: If you are just looking to confirm an existing sequence or are looking for SNPs or other changes, you should use the minimap2 algorithm in the Assemble menu. This needs you to supply appropriate references sequences so click on the Add Ref toolbar button and select one or more references sequence to use. These can be in any format MacVector can read. Then select the reference sequence(s) and the fastq file(s) and choose minimap2 to run the assembly. This can be very fast. Though, for large genomes, you will need a LOT of RAM.
There are other times when you might want to directly look at the reads, or sort them and just keep the longest reads. To do this in MacVector, create an Assembly Project but then use Add Seqs to import the data. This will try to import the reads as individual sequences, but, to avoid excessive memory usage, it caps the number of individual sequences at 10,000. So, if there are more than 10,000 sequences in the file, it will import the data as a file reference (just as if you had chosen Add Reads). You can use the SplitFastqFile.app utility (also in /Applications/MacVector/Applescripts/) to break up fastq files into smaller chunks. In the Assembly Project you can double-click to open an individual read, or sort by length and then select the reads you want to keep and save them as a fastq file by choosing File | Export Selected Reads To..
From September 2024 all active licenses of MacVector Pro were upgraded to MacVector Pro with Assembler at no additional cost at renewal time or when upgraded. If you haven’t yet been upgraded, please contact the MacVector Support Team!