
Tag: weeklytip
-
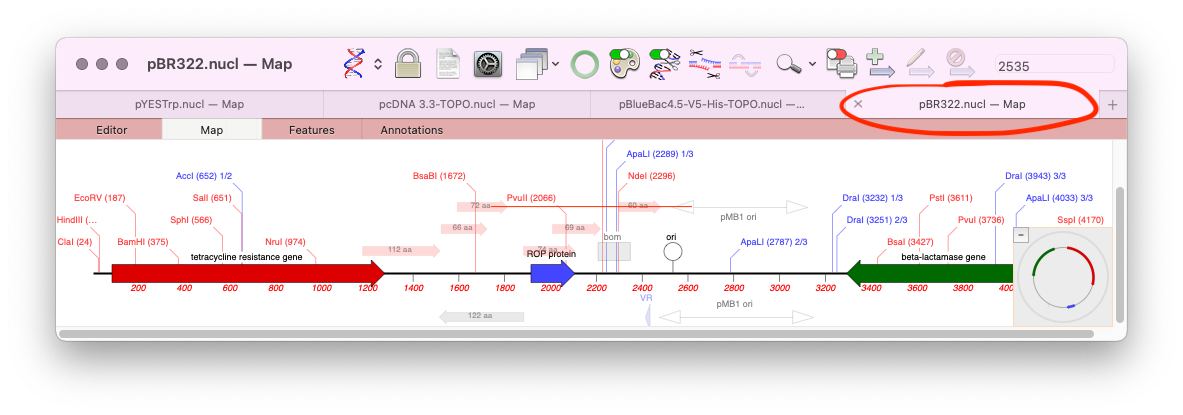
macOS’s tabbed windows and MacVector
One of the lesser known features of macOS is the ability to store all open documents of an application in tabs. Tabs were initially introduced for the Finder, but macOS Mavericks saw them apply to supported application document windows too. MacVector has supported tabs since their introduction, however, by default the Tab Bar is turned…
-
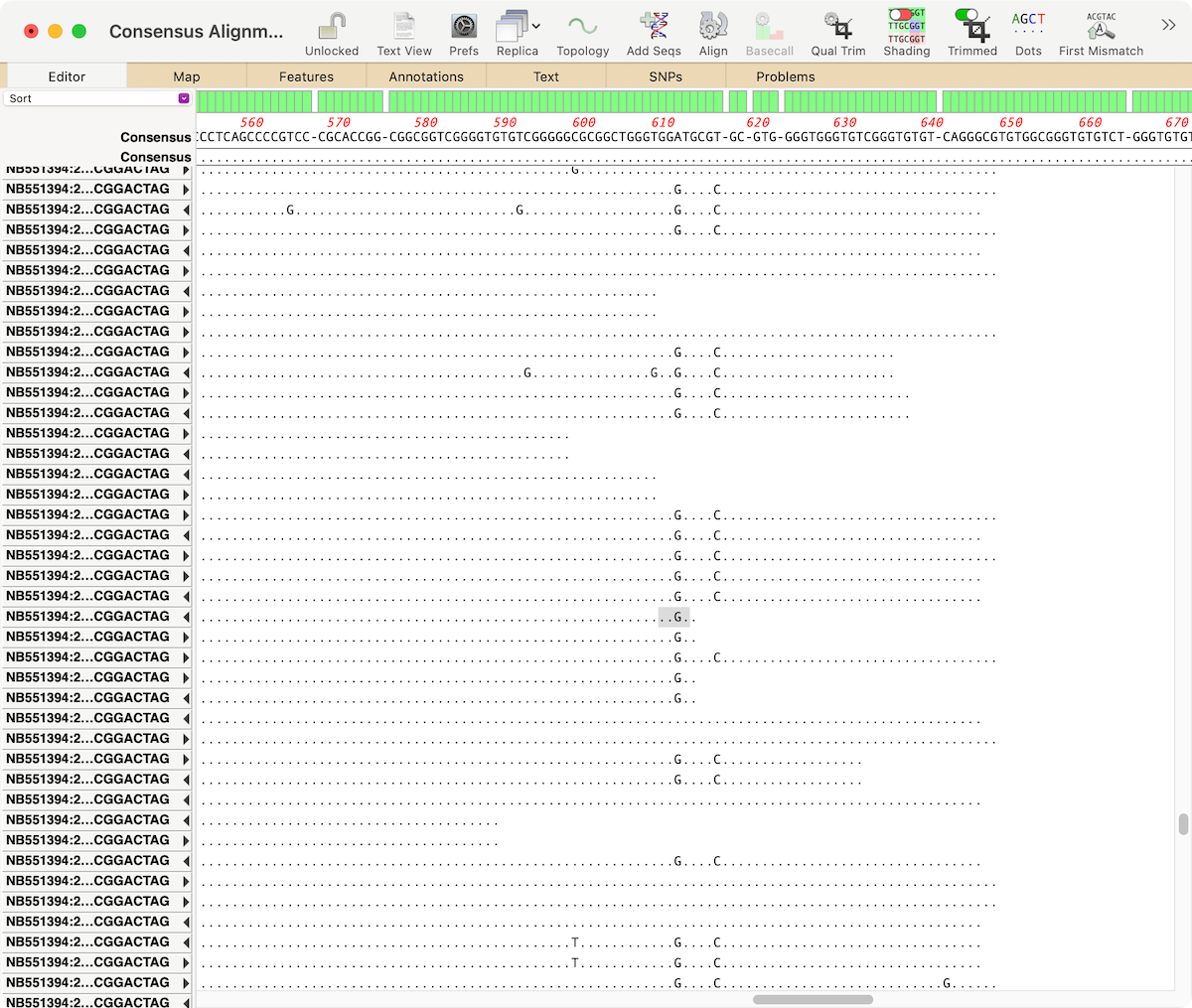
MacVectorTip: Identifying, Selecting and Assembling NGS reads with a variant genotype
When analyzing/assembling/aligning NGS data, there are many scenarios where you might want to separate out the reads representing different genotypes or variant sequences. MacVector makes this very easy. Take a reference sequence and choose Analyze | Align to Reference. Now click the Add Seqs button and select and add your NGS data files. NOTE: if…
-
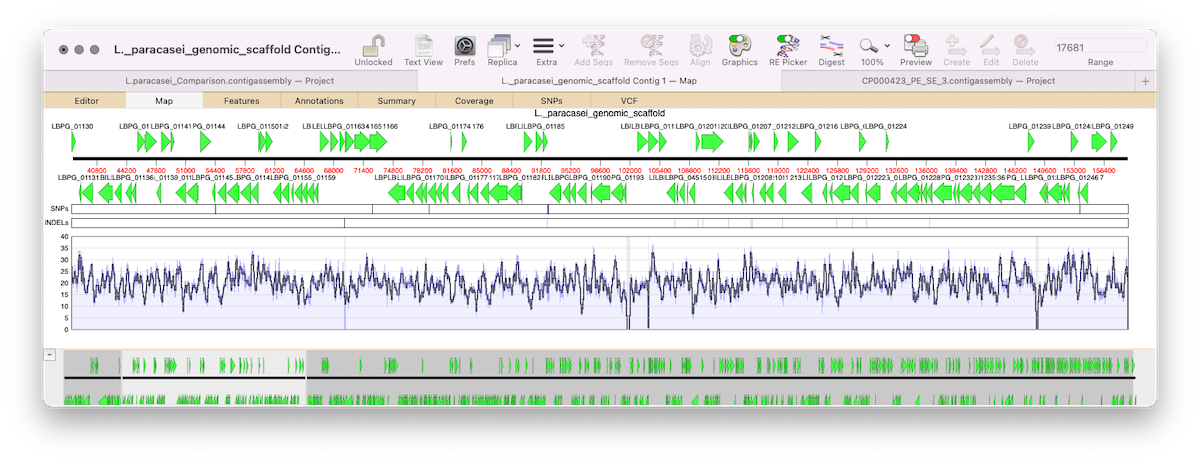
MacVectorTip: correctly flagging PacBio and Oxford Nanopore datasets for assembly by Flye
MacVector 17.5 introduced Flye for assembly of PacBio and Oxford Nanopore reads. Flye joins Phrap, Velvet and SPAdes for de novo sequence assembly using along with Bowtie2 and Align To Reference for reference assembly. Flye is an assembler algorithm tuned to assemble low quality long reads such as those produced by the new generation of…
-
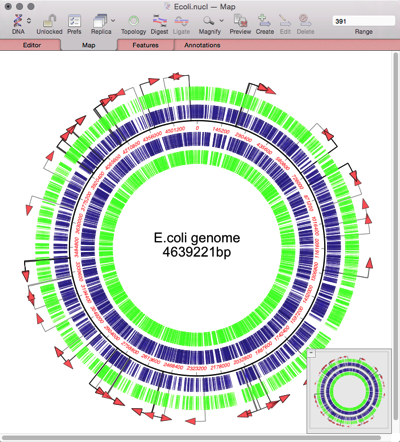
MacVectorTip: Assembling Fungal Genomes using SPAdes
MacVector with Assembler can assemble bacterial genomes in just minutes on quite modest hardware. Currently MacVector has four de novo assembly tools (SPAdes, Velvet, Flye and Phrap). But what of larger genomes? It is currently impractical to run de novo assemblies of Human genomes on a low cost Mac, though RNA-Seq analyses against the human…
-
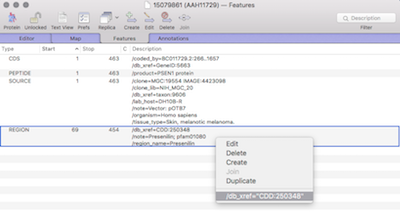
MacVectorTip: Viewing external database entries for features in a sequence.
Sequences, or regions of sequences, can be linked to external databases. For example an entire sequence entry or for when annotation tools are used to annotate proteins with domain or motif information (for example InterProScan). Very useful for when you want to view more detailed or updated information. Within the Genbank specification, which MacVector extensively…
-

MacVectorTip: How to Customize Window Button Toolbars
Like many Mac applications, MacVector takes full advantage of the built-in ability to add, delete and rearrange the action buttons on window toolbars. To make these changes, right-click (or [ctrl]-click) in the gray space on any toolbar and a context-sensitive menu will appear. Choose Customize Toolbar and a dialog will be displayed with all of…
-

MacVectorTip: Understanding Color Groups
You can align hundreds, or even thousands of protein sequences within MacVector using three different alignment algorithms – ClustalW, MUSCLE or T-Coffee. Once aligned, you may be familiar with the colorful display in the Editor tab. But there’s more to this than pretty colors. The default Color Group in MacVector is one called “Chemical Type”.…
-
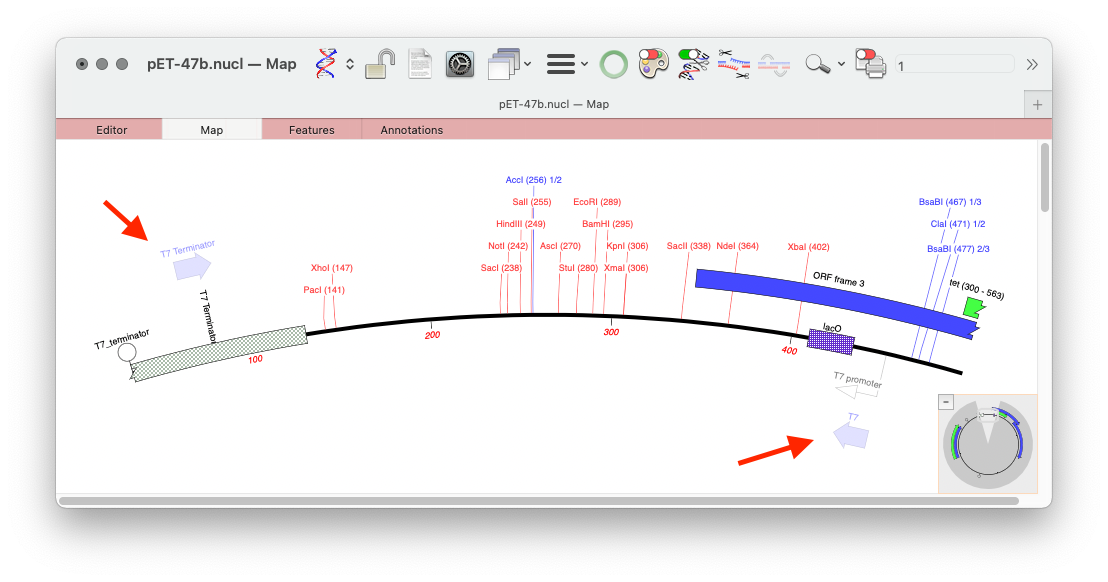
MacVectorTip: Scan For… Missing Primers: Automatically display Primer Binding Site on your sequences
MacVector’s Scan DNA For.. tool allows you to automatically display restriction enzyme recognition sites, putative ORFs, CRISPR PAM sites, missing annotation and also it will display primer binding sites from your own Primer Database in each DNA sequence that you open. Here’s an example of a couple of primers displayed on the pET 47b LIC…
-
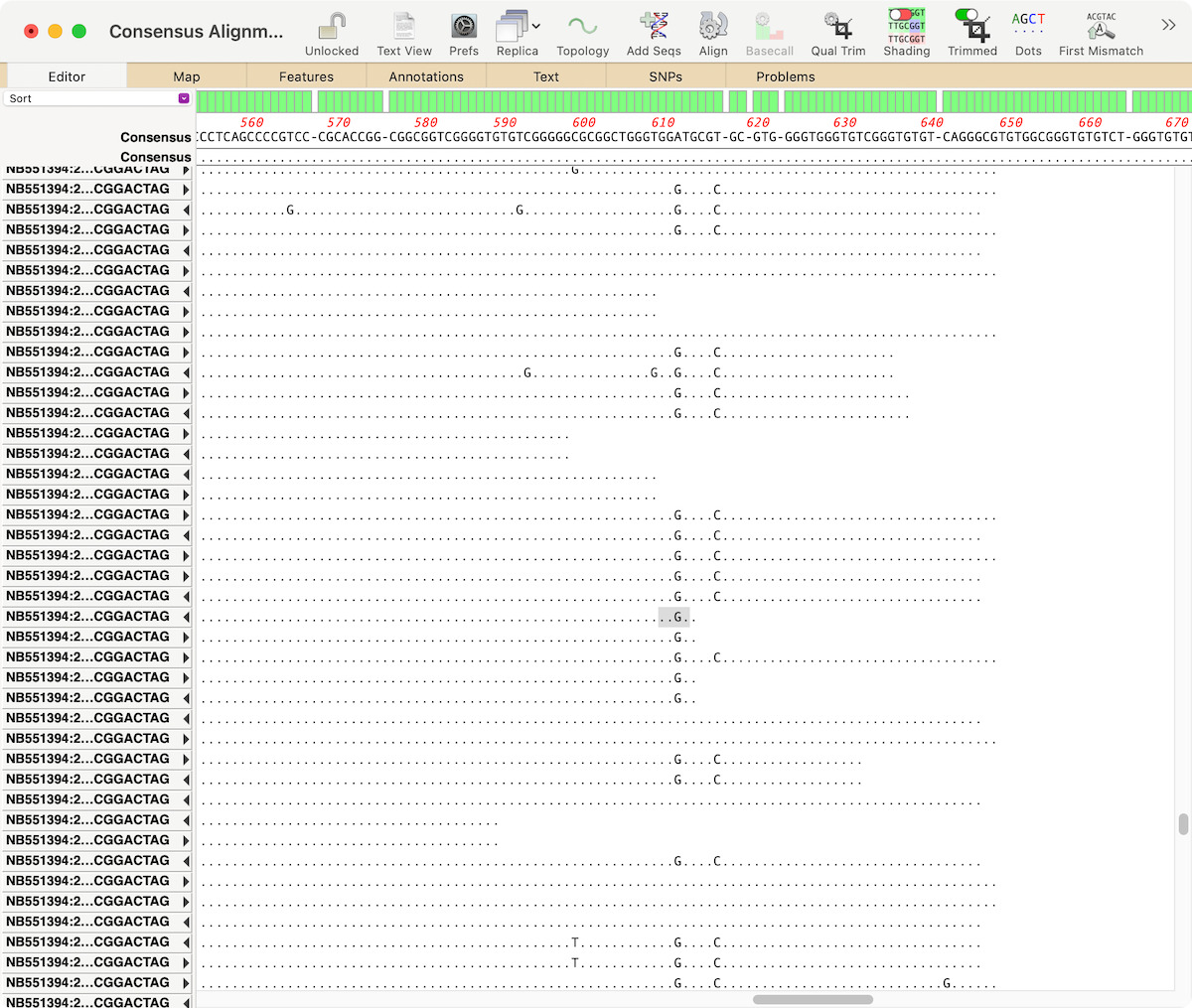
MacVectorTip: Viewing genotype changes in Align to Reference assemblies
The latest releases of MacVector, MacVector 18.0.1 (Intel) and MacVector 18.1.1 (Intel and Apple Silicon) have some tweaks to the output of the SNPs tab in the Align to Reference assembly window. The genotypes of any SNP changes now follow a consistent standard, and short deletions are also reported. If the region containing the nucleotide…
-
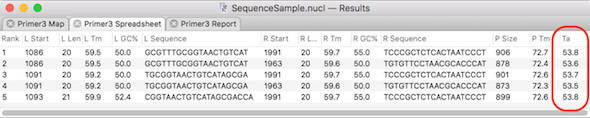
Calculating the optimal PCR annealing temperature
MacVector has several tools to help with primer design and testing. The Analyze | Primer Design/Test (Pairs) function uses the popular Primer3 algorithm to find suitable pairs of primers to amplify specified segments of DNA. You can also enter pairs of pre-designed primers and test their suitability for use in PCR. In both cases, the…