Multiple Sequence Alignment
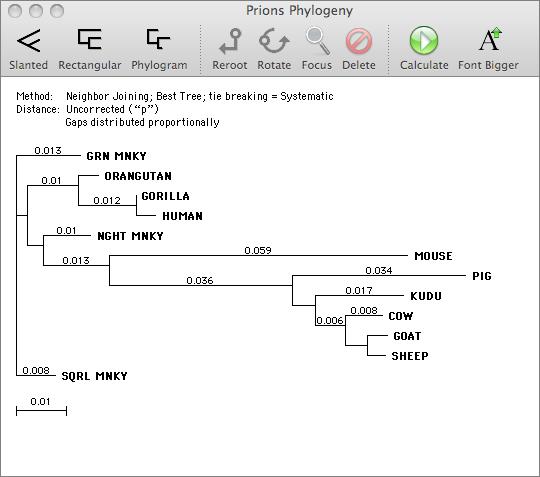
You can use MacVector to align related DNA or Protein sequences. MacVector uses the ClustalW algorithm to automatically align any number of sequences and provides a sophisticated editor that lets you fine tune the alignments. The alignments can be output in a variety of formats or used to generate phylogenetic reconstructions using a number of built-in algorithms.
Creating Alignments
You can create alignments from sequence already open in MacVector, or by creating an empty alignment and populating it with sequences imported from disk. You can also directly open an existing file containing multiple sequences. MacVector supports the following multiple sequence file formats:
MacVector
PHYLIP
NEXUS
FastA
GCG MSF
GCG RSF
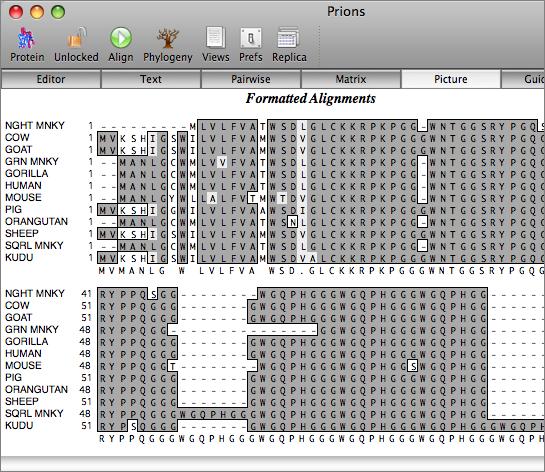
You work with alignments in the multiple sequence alignment editor. Sequences can be viewed using plain monochrome text or colored according to user-defined color groups.
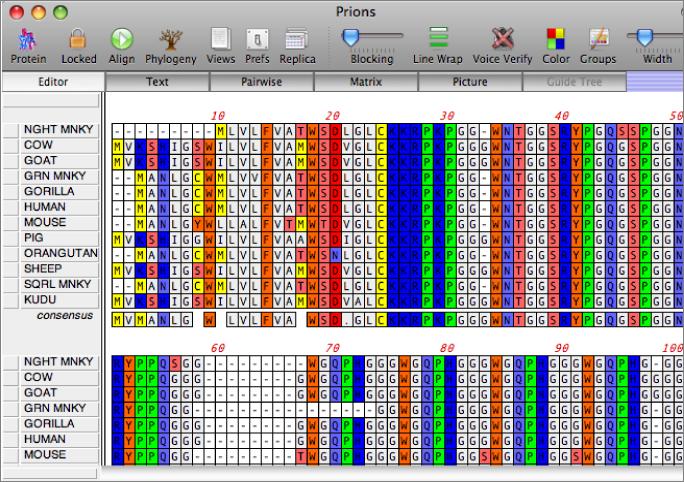





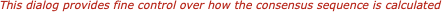
There are a variety of parameters and modes that control how the residues appear on-screen. For example, you can set up the editor so that all matches to the consensus are displayed as dots - this is very useful for identifying variant regions in otherwise closely related sequences.
Output Options
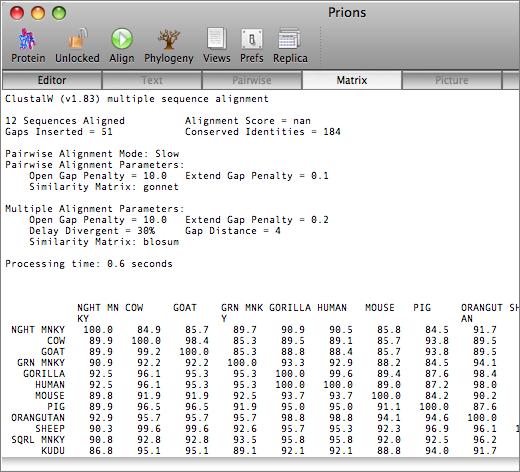
The aligned sequences can be displayed in result windows in a variety of different ways. One window provides a high quality customisable graphical text display designed for output on a laser printer, while another duplicates the output from ClustalW. You can also view the sequences aligned in pairwise mode, along with a matrix displaying the identities and similarities of each pair of sequences.





|

