What's New in MacVector 13.5?
Overview
The graphics Symbol Editor and floating Graphics Palette have been rewritten in preparation for MacVector moving to a 64-bit architecture (due with MacVector 14.0). The other main enhancements have been aimed at better handling of Next Generation Sequencing (NGS) files, particularly with the Align To Folder and Assembler Velvet de novo assembly functionality. There has also been significant code optimization to better handle the analysis of large genomic sequences, particularly noticeable with the Pustell Matrix “dot-plot” functionality.
Align To Reference Enhancements
The Align To Reference alignment algorithm and the Editor now handle alignments around a circular sequence. Note that this is only the case for the Sequence Confirmation algorithm. The cDNA Alignment algorithm still assumes the target sequence is linear.
You can now select one or more sequence “Reads” in the Align To Reference Editor and save those reads to a fasta or fastq formatted file by choosing File | Export… and selecting the required format in the resulting dialog. The primary benefit of this new feature is that you can now run alignments of up to 500,000 NGS reads against a reference sequence and then specifically export all of the aligned (or non-aligned) reads for further analysis.
Align To Folder Enhancements
Align To Folder can now perform alignments against fasta and fastq files with many millions of reads. On a relatively new laptop, a search of a 1kb sequence against a ~2.5 MB fastq file containing 10 million 100nt reads takes about 40 minutes.
You can now retrieve the “hits” from an Align To Folder run to one of three destinations – the MacVector desktop (i.e. opening each hit in a window on the screen), to a folder (where each hit is written out as a separate file) or to a single file (where the hits are concatenated into a single fasta or fastq file). The key to this functionality is that you can select lines in the Folder Description List window as follows;
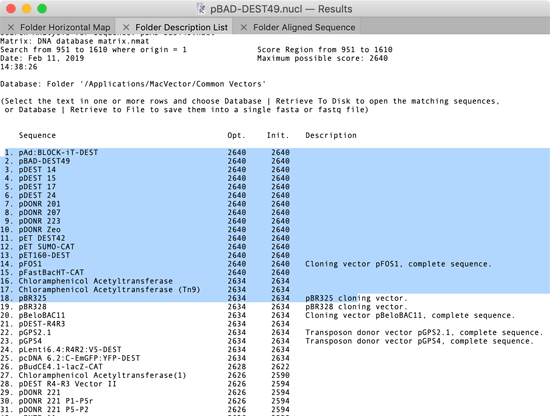
When any part of a line is highlighted, that “hit” is considered “selected”. You can then choose one of the options from the Database menu;
Database | Retrieve To Desktop – this opens windows on the desktop containing the selected sequences. If the matching sequence was a single file (e.g. in MacVector or GenBank format) the original file is opened. If the hit is a Read within a large fasta or fastq file, a new sequence window is created and populated with the Read sequence.
Database | Retrieve To Disk – this saves the hits to individual files in the selected destination folder. If the matching sequence was a single file (e.g. in MacVector or GenBank format), the file is simply copied to the destination so that all features and feature appearance information is maintained.
Database | Retrieve To File – this saves the sequence information (plus any quality information in the case of fastq data) of all selected hits into a single file in either fasta or fastq format.
Taken together, these enhancements let you use Align To Folder to pull out rare matching reads from large NGS datasets for use in further analysis or DNA Assembly. In beta testing, this has been used to “clone” genes from MiSeq RNA-Seq runs using Protein source sequences aligned to fastq NGS data.
Pustell Matrix (“Dot Plot”) Enhancements
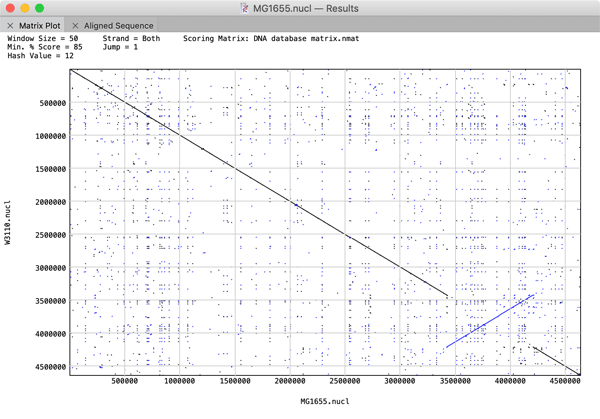
The performance of large (i.e. genome-sized) Pustell Matrix dot plot alignments has been dramatically improved. You can now scan and display pairs of bacterial genomes in just a few seconds to easily identify inversions, duplications and rearrangements in their gene organization. You can zoom in and out of the dot plot display in real time to explore the relationships right down to the residue level. If you are using large sequences, consider increasing the Hash Value, Window Size and Min % Score values to speed up the calculations. For example, the plot below comparing two E. coli genomes was generated in just a few seconds using Hash Value=12, Window Size=100 and Min % Score=85.
Nucleic Acid Subsequence Enhancements
A small but significant change has been made to the graphical output of this. If you select a pair of hits in the Map results tab, then choose Edit | Copy, the sequences of the actual subsequences are substituted into the copied sequence. This allows you to maintain primers in a nucleic acid subsequence file and use the Nucleic Acid Subsequence analysis option to quickly identify and “clone” predicted PCR fragments even if the primers have mismatches or tails added to them.
Assembler Enhancements
Reference alignments (from Bowtie) now have a separate coverage report tab that lists the read coverage for every gene and CDS feature in the reference sequence. You can use this to (e.g.) accurately measure relative expression levels of mRNA in RNA-Seq experiments or plasmid copy number in whole cell DNA sequencing experiments.
Velvet now does a much better job at trimming poor quality residues from input sequences.
Velvet now handles paired sequences that have identical names.
You can now export unassembled reads from Bowtie and Velvet alignments as fasta or fastq files. This lets you filter out reads that match specific sequences so you can focus subsequent alignments using a subset of reads.
Miscellaneous Enhancements
You can now reset the circular origin of circular sequences to any arbitrary position. Simply click between the residues where you want the new origin to be, then right-click (or <control>-click) and choose Set Circular Origin from the popup menu.
Informative tooltips have been restored in all views. There is a setting to turn this on/off in MacVector | Preferences | General.
The cDNA Align To Reference algorithm now does a much better job of displaying segmented reads in the Map view.
A number of issues with saving trace files have been resolved. In particular, you can now use the bsml format to save annotations with chromatogram information.
|

