
Author: Chris
-
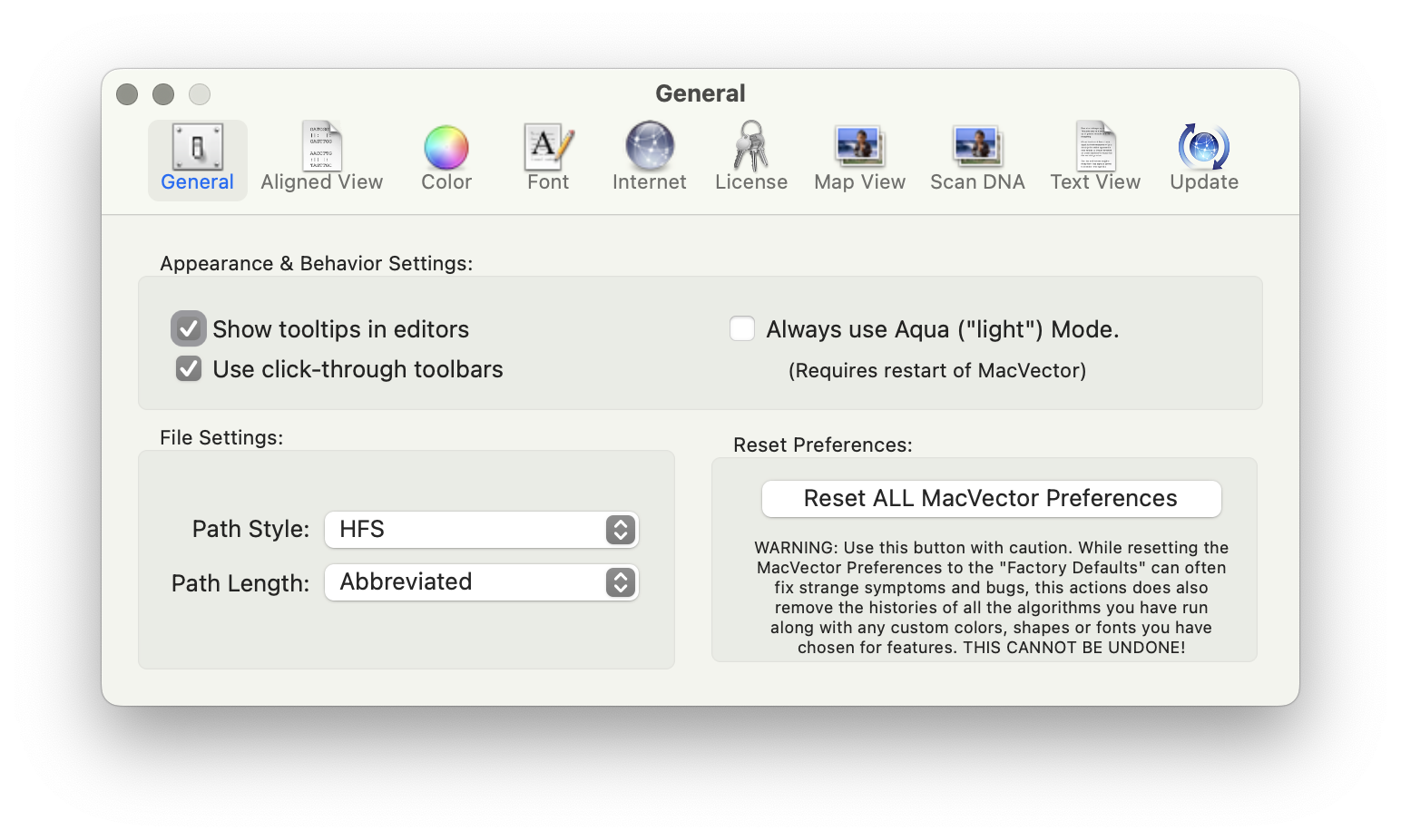
MacVectorTip: MacVector 18.8 has a new Preference Reset button.
Very occasionally, you may find MacVector behaving unusually. Perhaps windows are not activating correctly, or functions you have used without issue for many years have suddenly started crashing, taking an excessive amount of time to run or generating nonsensical results. When such issues are reported to MacVector Support, we always try to perform an in-depth…
-

MacVectorTip: Use Quicktest Primer to design primers that change encoded amino acids and create restriction enzyme sites
The Quicktest Primer interface is highly interactive and the display shows restriction enzymes and the amino acids sequences of CDS features in the region of interest. Here’s a ~25nt primer aligned against a parental sequence. Restriction enzyme recognition sites are shown in black text. “One-out” sites (e.g. a 5 out of 6 match) are shown with an…
-
MacVectorTip: Make Sure You Have Assembler Activated!
In September of 2024, we started including Assembler with all new, upgraded and renewed licenses of MacVector. Assembler is fully integrated within MacVector and is enabled by the license activation code. However, it may be that your lab or institution has a new Assembler activation code after a renewal, but you are still using an…
-
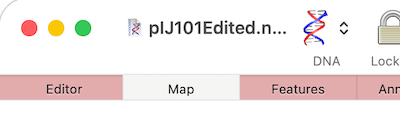
MacVectorTip: How to Control the Length of Window Title
If you are having difficulties viewing the name of your sequence in the title bar of a window, it may be because you have “Full Titles” turned on. So if your window title looks like this; Then open MacVector’s Windows menu and deselect Show Full Titles. …and you now just see the name of the…
-

MacVectorTip: Assembling Nanopore or PacBio Long-Read Data with Flye
In the previous post we discussed the various ways in which you can analyze Oxford Nanopore’s long read data. For de novo assembly we recommend using Flye, which can also be used with PacBio data. Here are some tips to get the most out of Flye. IMPORTANT: MacVector simply wrappers around the Flye executable algorithm which depends on…
-
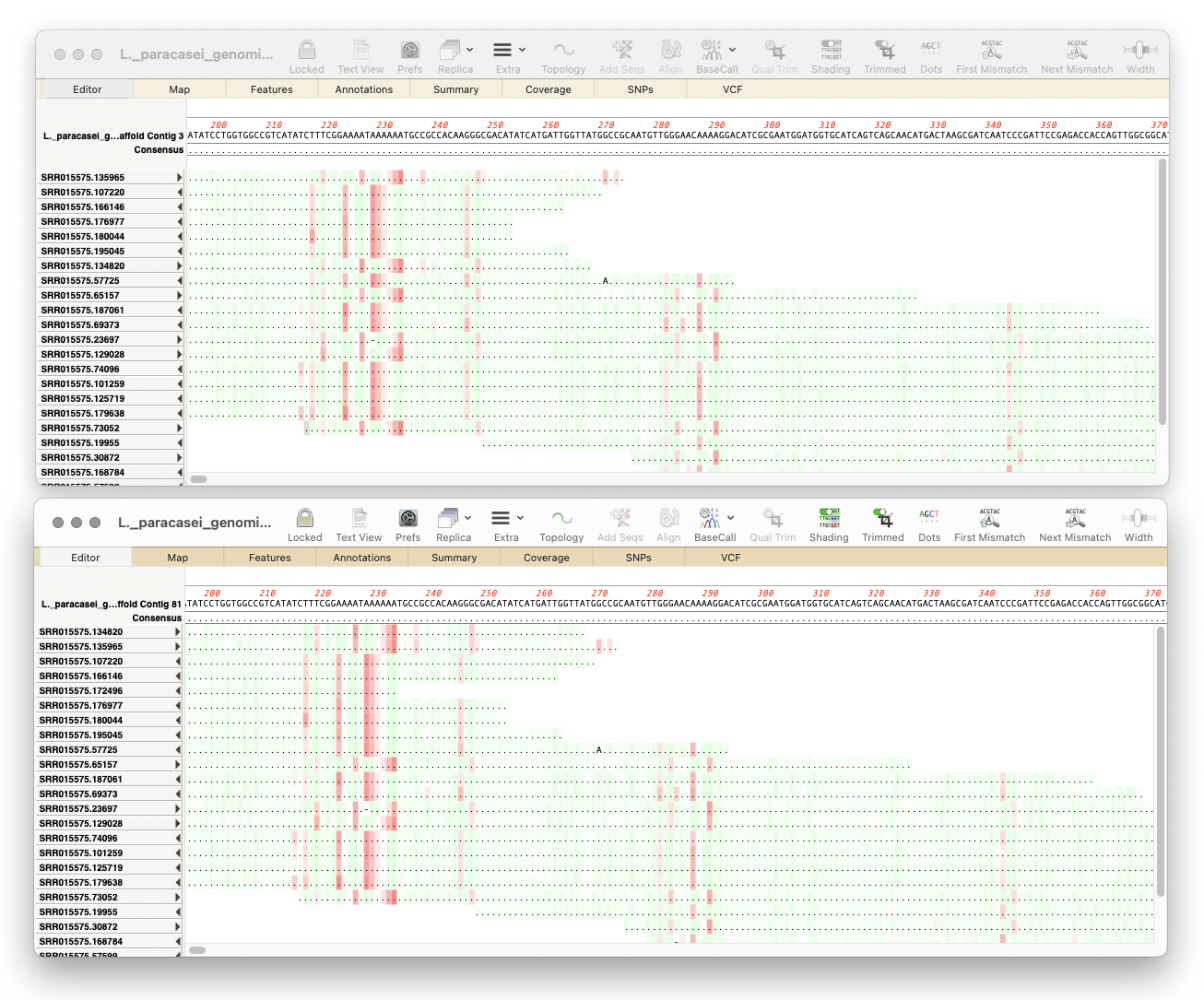
MacVectorTip: Working with Oxford Nanopore Long Read Data
Here’s a few tips regarding analyzing long read data from the Oxford Nanopore Technology MinION and GridION machines. First, you should always first create a File | New | Assembly Project and then (typically) click on the Add Reads toolbar button and select the appropriate fastq formatted data files. These are often supplied in compressed…
-
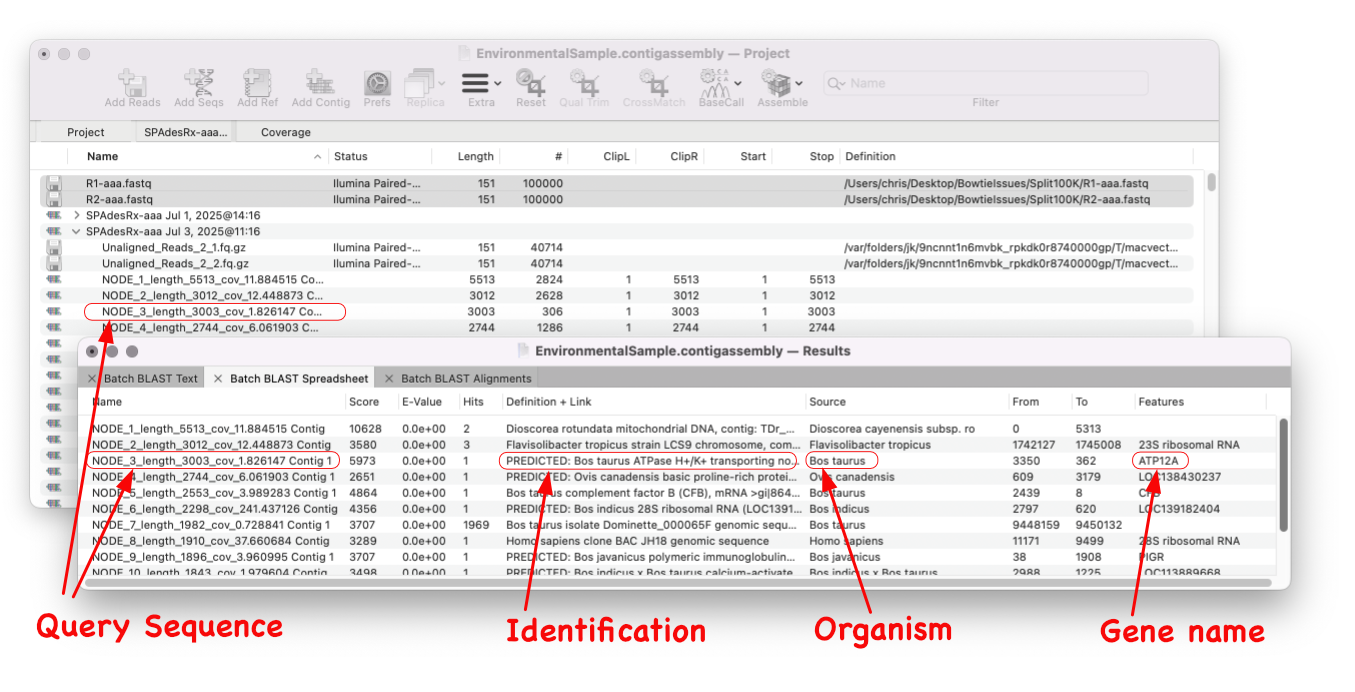
Batch BLAST
MacVector 18.8 is out and it’s packed with new tools! MacVector 18.8 has tools to help you identify and annotate unknown, unannotated or partially annotated sequences. Ideal for identifying contigs from a de novo assembly. One of these new tools is AutoAnnotate (via BLAST) Batch BLAST is a game-changing feature that revolutionises the way you identify unknown sequences. With…
-
Merry Christmas from the MacVector team
For those who celebrate the MacVector team wish you a very merry Christmas. We hope you are able to turn off that sequencer, put away your pipette and have a relaxing time with your loved ones. Here’s to a better year for science in 2026!
-
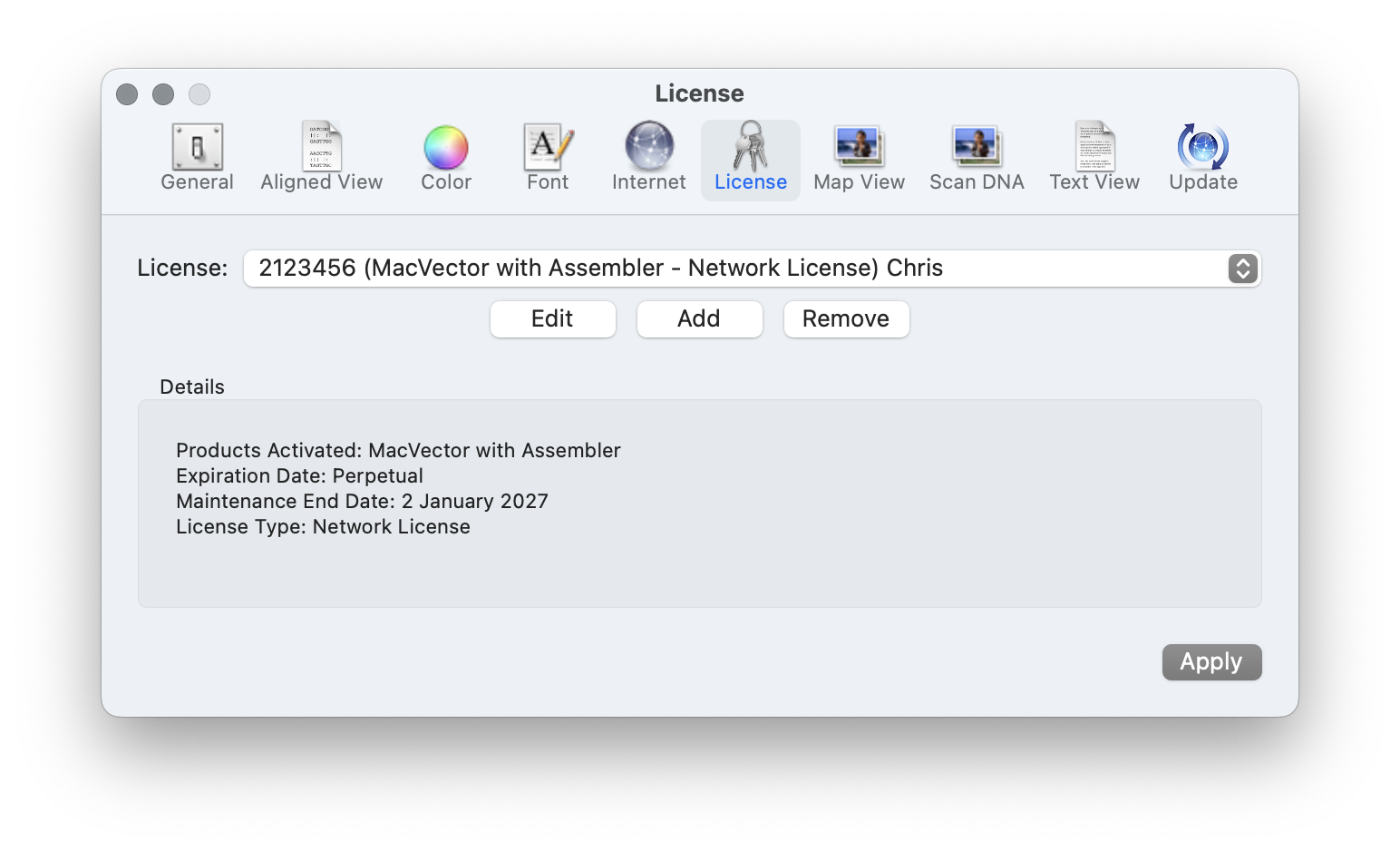
Migrating to a serverless concurrent usage license
As well as having a license per user for larger sites we have concurrent usage or network licenses that allow an entire institute, or a large research group to share MacVector. Concurrent usage licenses give you a pool of licenses that can be shared across an entire network. The number of seats defines how many…
-
MacVector 18.8.2 is out
We’ve just released a minor update to MacVector 18.8. The main change is improvement to Codon Bias Table (CUT) import. But there are multiple bug fixes too. The release notes contain full details. You should be prompted to upgrade in MacVector, but you can also upgrade immediately: MACVECTOR | CHECK FOR UPDATES… You can also…