Even the latest Macintosh computers loaded with as much RAM as you can afford will still struggle to de novo assemble genomes much over 50 Mbp. But, often that is not required. If you are just interested in a few genes, or a specific region of a chromosome, you can use Align to Folder to filter the reads and save just those reads that you are interested into much smaller fasta/q files and easily manipulate those in MacVector.
Here, we used the ~2kb cdt operon from Campylobacter jejunii NCTC 11168 which encodes “cytolethal distending toxin”, an important pathogenicity determinant, and wanted to find and compare the same operon from the related C. jejunii ATCC 35925. The source data was a pair of gzip’d MiSeq genome DNA read files each containing 770,000 x 300nt reads.
The Align to Folder search was set up as follows;
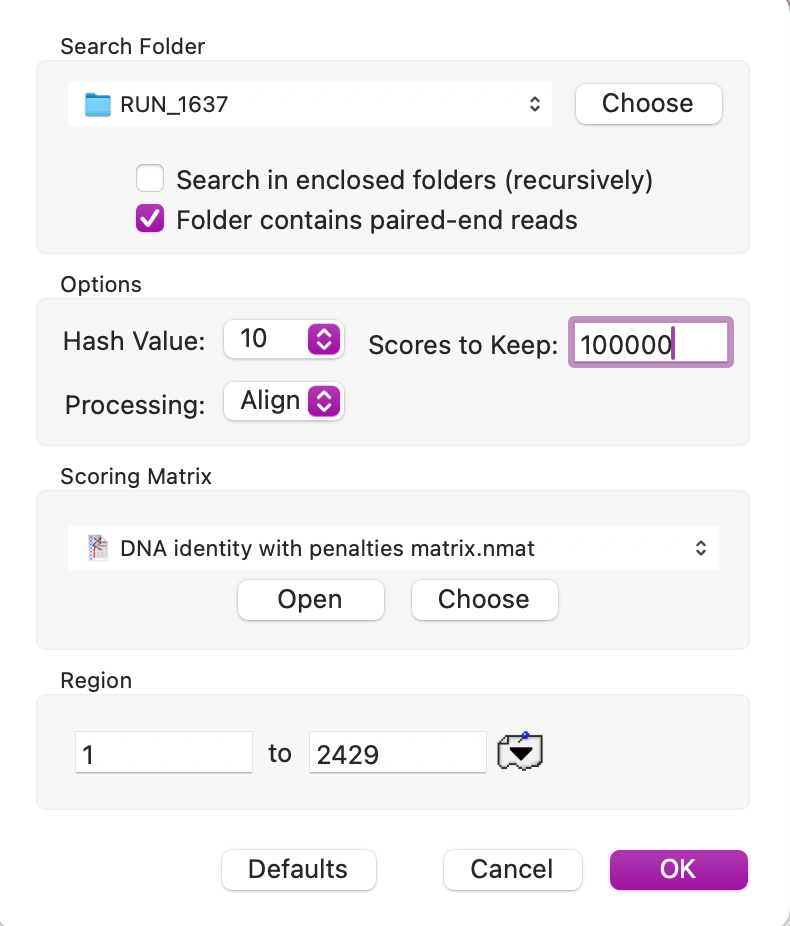
The search took 8 minutes on an M1 MacBook Pro. Once complete you can select the hits in the Description List output;
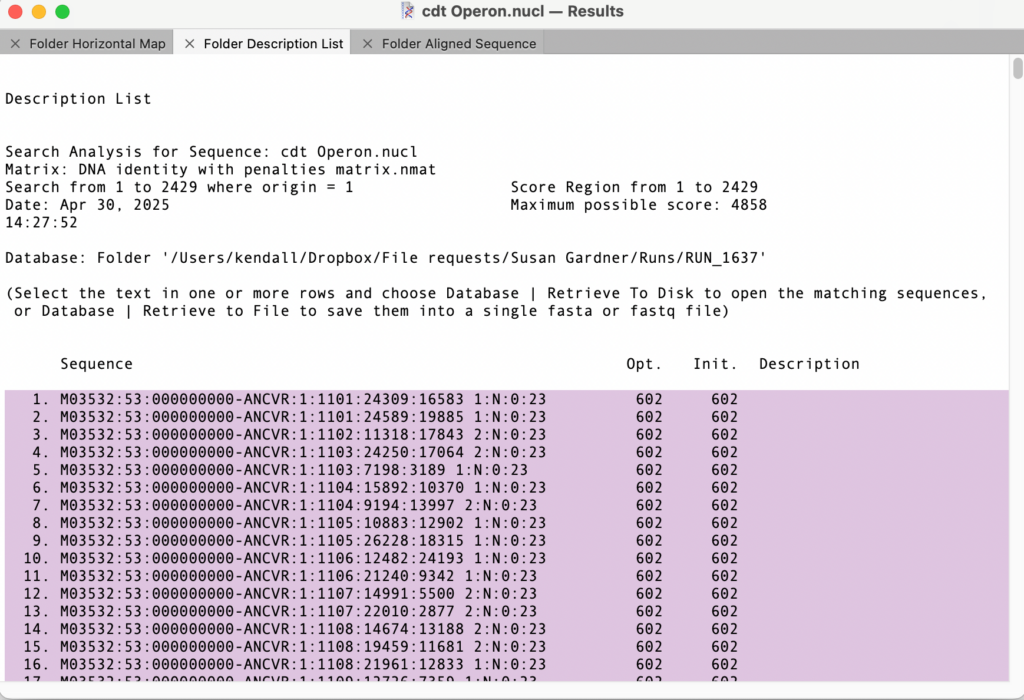
You have a choice here – you can carefully select from just before the first sequence, scroll towards the end and select just after the last hit that you feel is valid, or “select-All” to select everything. Then choose Database | Retrieve to File to save the hits to a new file(s). If the source data were paired end files, two files will be created – any matching read will always have its pair included, even if that pair’s sequence did not match the query sequence. Note that this is a great way to extend the filtering of a sequence of interest by using the pair relationship to retrieve adjacent sequences.
Once you have the filtered data set, you can use many different approaches, all of which should complete very quickly.
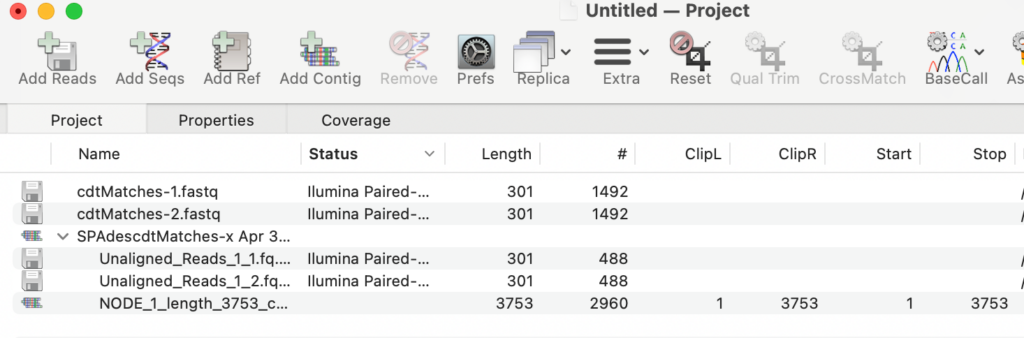
(a) Add the paired files to a new Assembly Project and assemble with SPAdes (short reads) or Flye (long reads). For this example, most of the ~3,000 matches assembled into a single 3,753nt contig which can then be used in other MacVector analyses
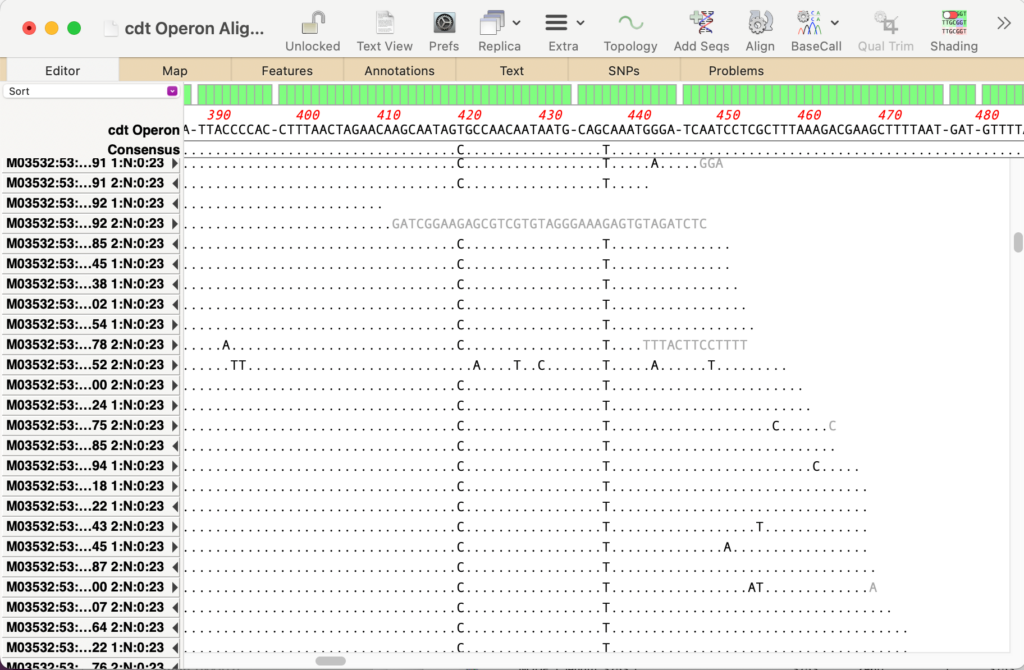
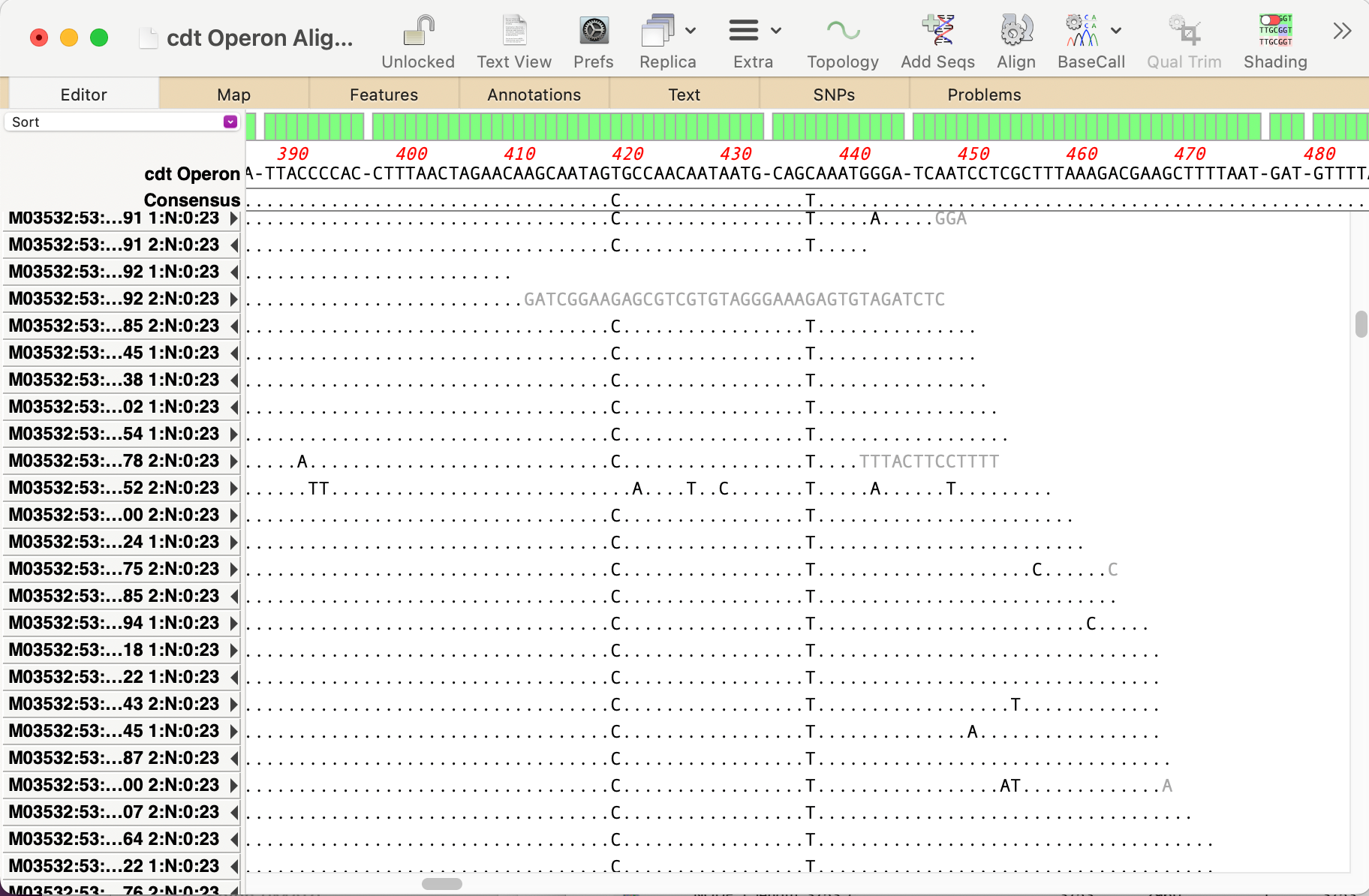
(b) You can run an Align to Reference using the original operon as the reference and adding both filtered fastq files via the Add Seqs toolbar button. After running Align, SNPs can be easily seen.