MacVector has a wide array of different tools for working with protein and DNA sequences. MacVector has always been designed with the Mac’s simplicity in mind and getting started with simple tasks is quick. However, making the most of the many functions and getting familiar with MacVector’s wide range of tools does require more help…
MacVector 17’s brand new Restriction Enzyme Picker gives you an interactive way to quickly show what enzymes will digest your sequence. You can dynamically filter what sites are displayed. For example to show just 3’ overhang cutters or use other criteria. What’s more is that you can take a set of cut sites from one…
MacVector 17 will be released later this week. Get ready for our biggest release yet. MacVector 17 compares genomes, makes restriction enzyme cloning easier, automates the design of Gibson Assembly and Ligase Independent Cloning strategies. Makes plasmid maps even more beautiful and supports macOS Mojave’s Dark Mode to aid concentration on those late night primer…
One new feature in our MacVector 17 release is the ability to automatically display primer binding sites in each DNA sequence that you open. Here’s an example of a couple of primers displayed on the popular pET 47b LIC cloning vector on each side of the LIC cloning site. The image shows how MacVector 17…
Macs are pretty good at choosing the right application to open a document. For example when you double click on a .nucl document then it will open in MacVector. However, sometimes this file association breaks. Applications should coexist peacefully on a Mac, but sometimes a misbehaving app will corrupt these file associations and you will…
If you are interested in making changes to a protein sequence, it is often useful to make a change to the coding DNA that will create a restriction enzyme site. Conversely, there may be times that you would like to create a site without affecting a protein coding region. You can accomplish this using the…
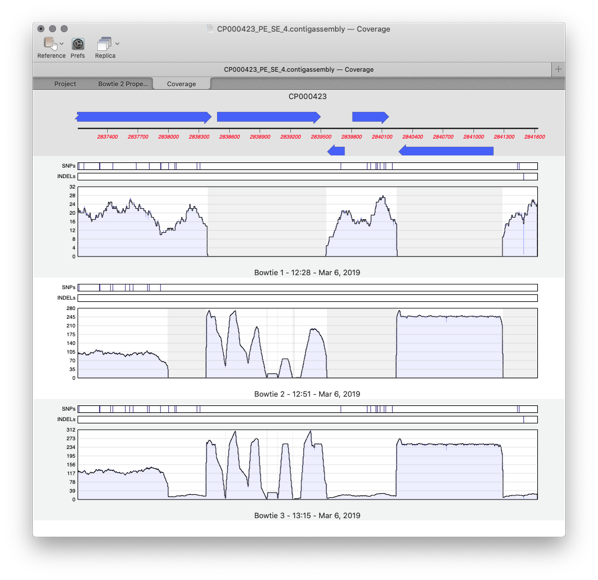
MacVector 17 has a greatly improved Assembly Projects manager, for better organization of multiple sequencing datasets, multiple references sequences and repeated jobs. Every time you run a new assembly job (either a reference assembly or de novo). A new job object is created in the Assembly Project window contains resulting contigs and any unaligned reads…
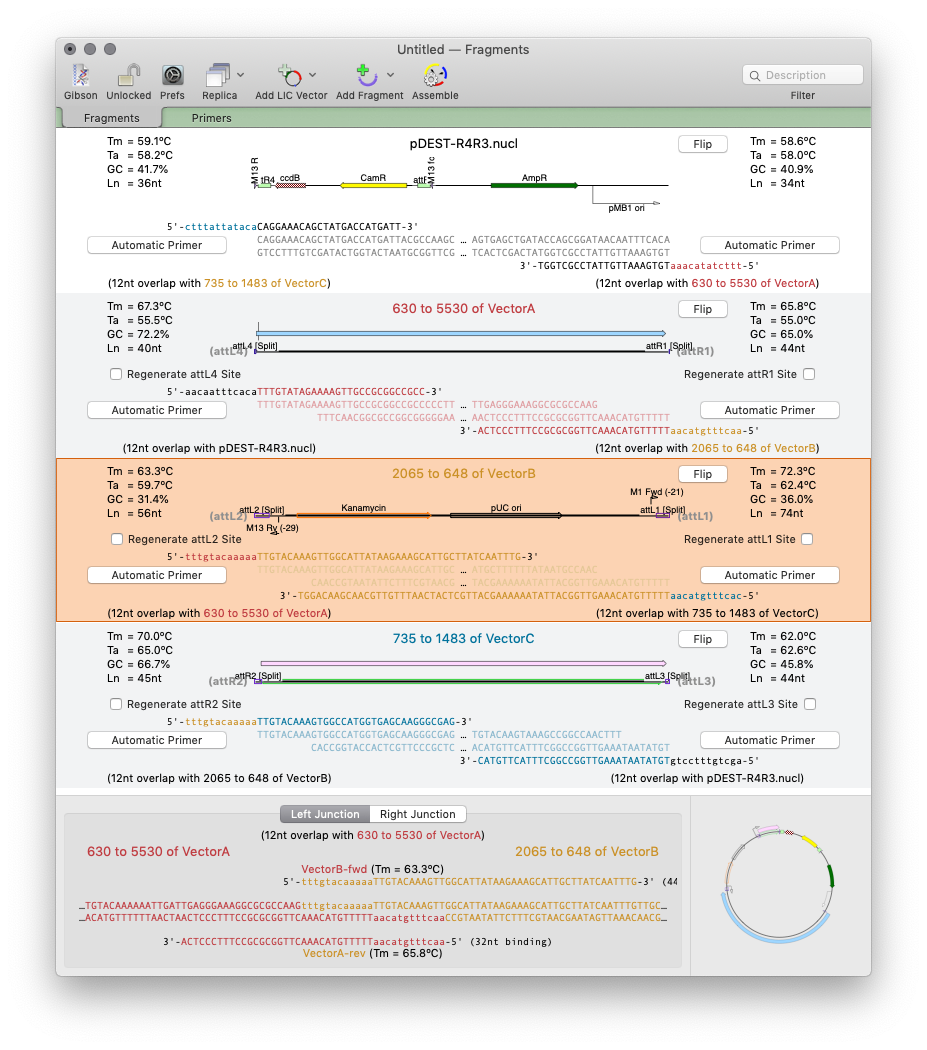
MacVector 17 has a completely new tool for automated design of ligase-independent cloning strategies. The tool supports 5’ exonuclease driven Gibson assembly as well as the T4 DNA Polymerase 3’ exonuclease “Ligase Independent Cloning” approach. MacVector can automatically design primers when you specify fragments and vectors to use. You can provide custom primers (manually or…
We recently became aware of a few issues with Entrez and BLAST. The most common issue is that in Entrez there are only two databases that you can search. The main cause is due to some changes the NCBI made to their service, that went live on the 1st of December, 2018. During the implementation…