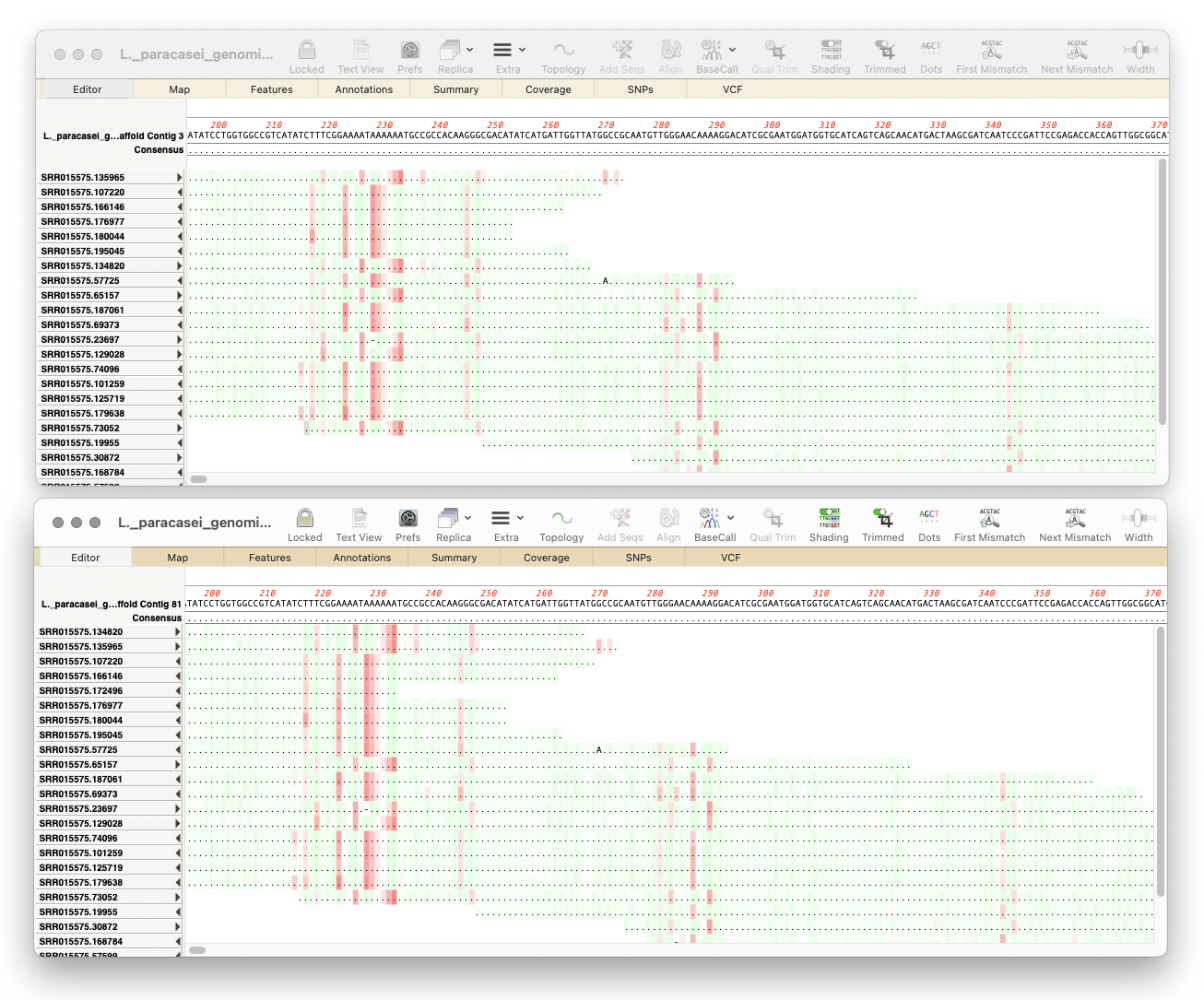
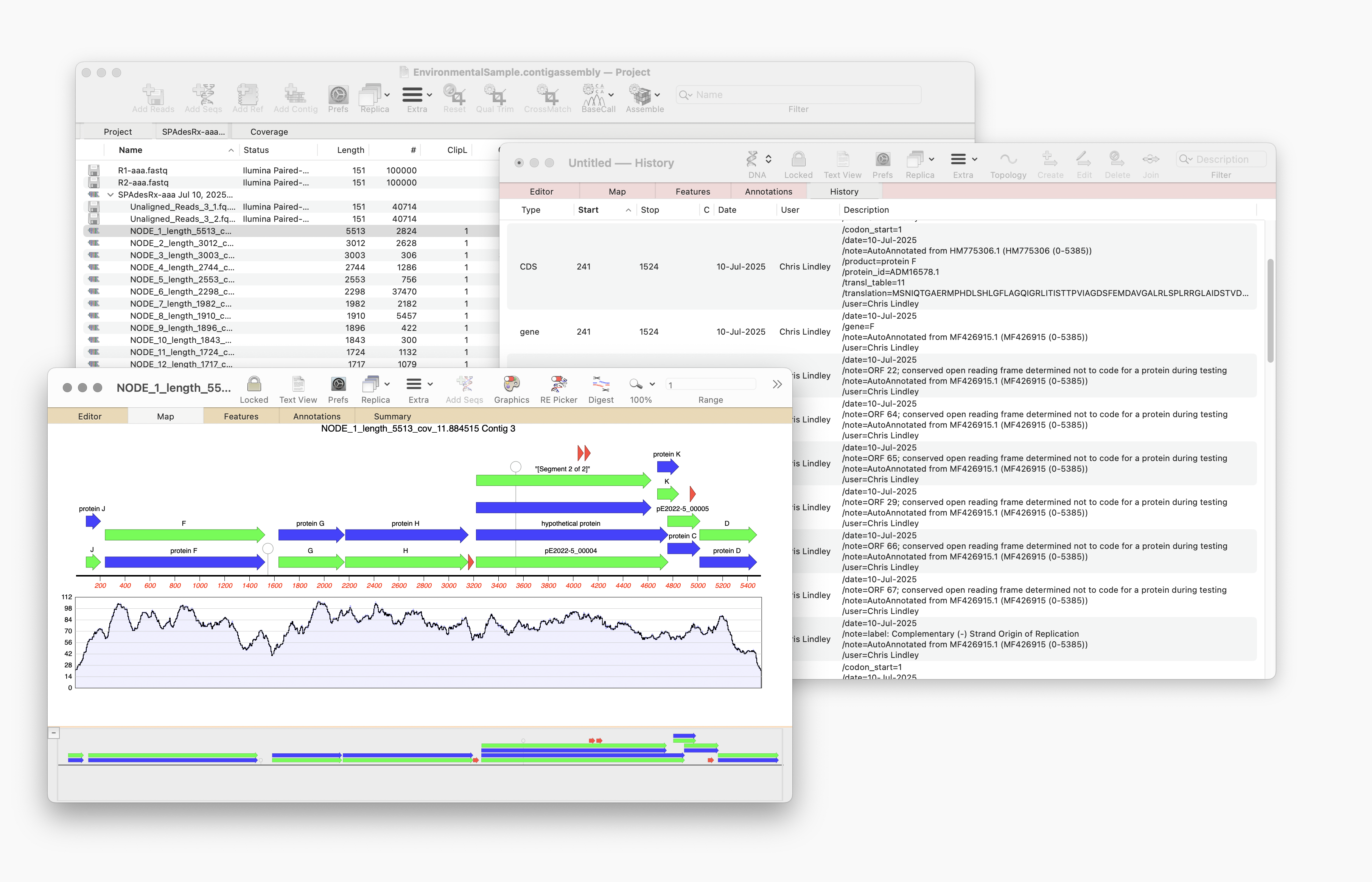
Here’s a few tips regarding analyzing long read data from the Oxford Nanopore Technology MinION and GridION machines. First, you should always first create a File | New | Assembly Project and then (typically) click on the Add Reads toolbar button and select the appropriate fastq formatted data files. These are often supplied in compressed…
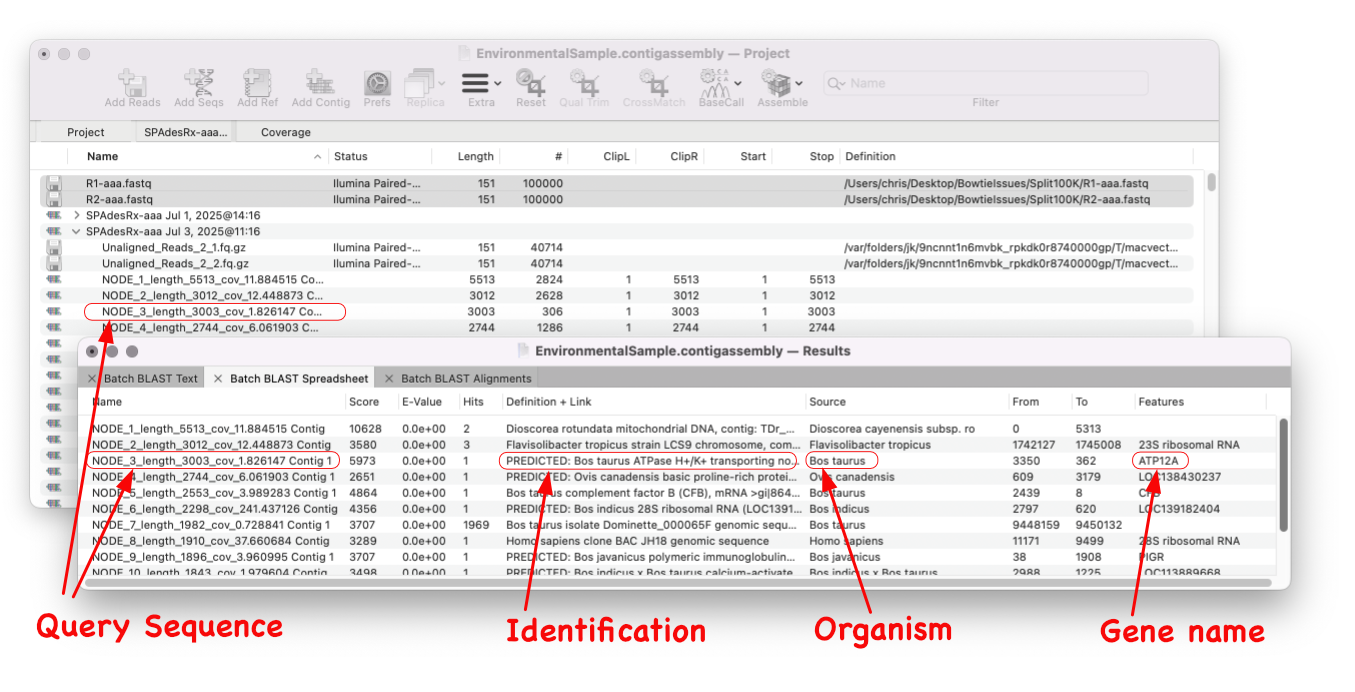
MacVector 18.8 is out and it’s packed with new tools! MacVector 18.8 has tools to help you identify and annotate unknown, unannotated or partially annotated sequences. Ideal for identifying contigs from a de novo assembly. One of these new tools is AutoAnnotate (via BLAST) Batch BLAST is a game-changing feature that revolutionises the way you identify unknown sequences. With…
For those who celebrate the MacVector team wish you a very merry Christmas. We hope you are able to turn off that sequencer, put away your pipette and have a relaxing time with your loved ones. Here’s to a better year for science in 2026!
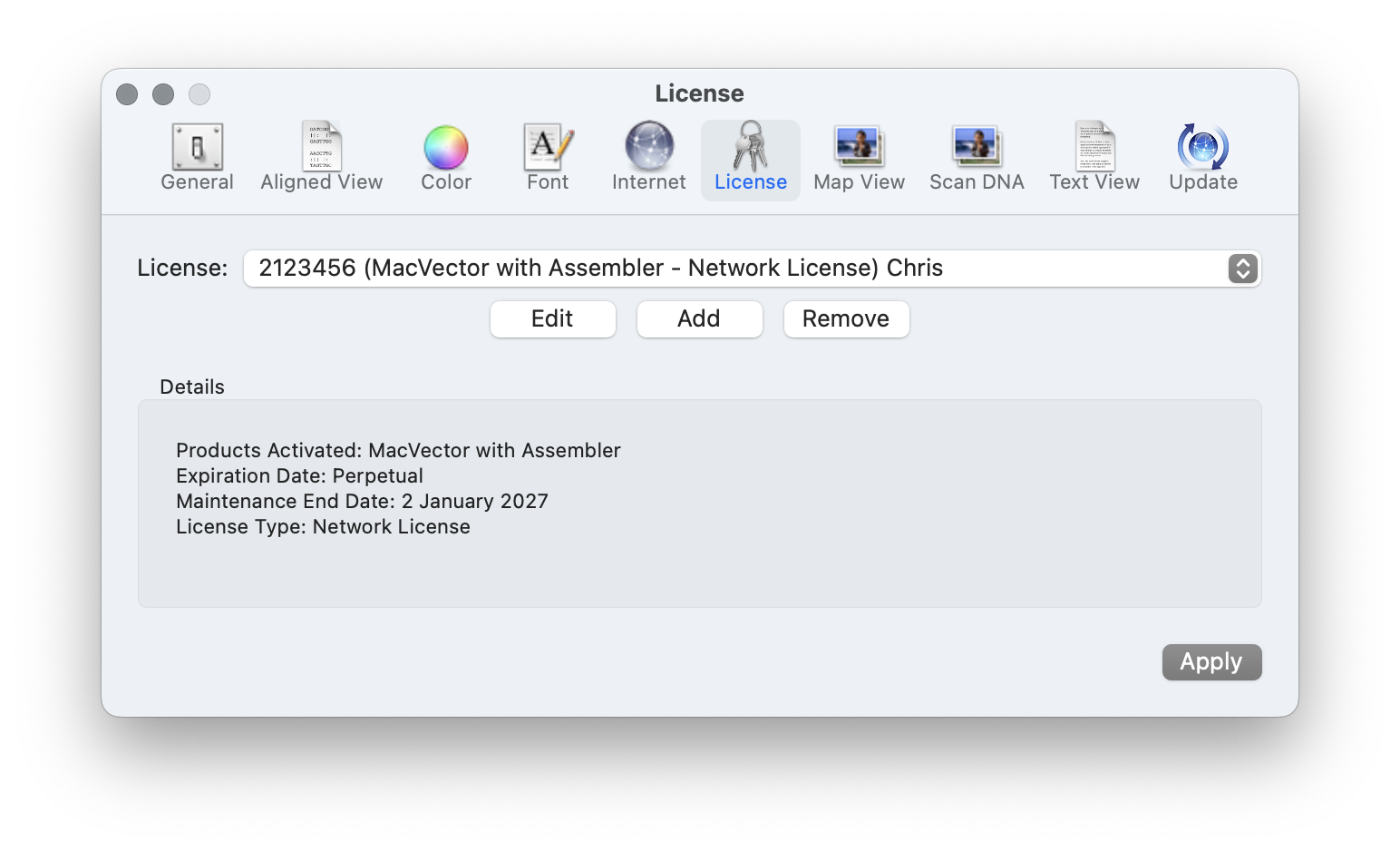
As well as having a license per user for larger sites we have concurrent usage or network licenses that allow an entire institute, or a large research group to share MacVector. Concurrent usage licenses give you a pool of licenses that can be shared across an entire network. The number of seats defines how many…
We’ve just released a minor update to MacVector 18.8. The main change is improvement to Codon Bias Table (CUT) import. But there are multiple bug fixes too. The release notes contain full details. You should be prompted to upgrade in MacVector, but you can also upgrade immediately: MACVECTOR | CHECK FOR UPDATES… You can also…
Apple released macOS Tahoe on Monday (15th Sept 2025). As usual in the run up to a new macOS release, we have been testing MacVector 18.8 on development and public betas of macOS Tahoe. With macOS Tahoe’s Liquid Glass appearance we have come across a very minor display glitch. But otherwise we are happy to…
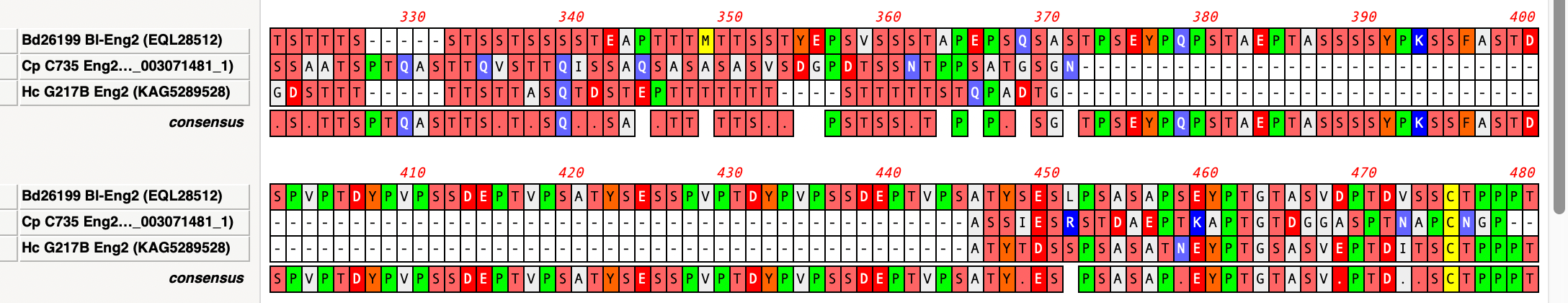
By default, MacVector ignores gaps when calculating the consensus of a multiple sequence alignment. However, this can lead to some unexpected results. For example, consider this three sequence alignment where one sequence has a long insertion compared to the other two. In this case it does not seem reasonable to believe that the “consensus” should…
MacVector 18.8 is out and it’s packed with new tools! MacVector 18.8 has tools to help you identify and annotate unknown, unannotated or partially annotated sequences. Ideal for identifying contigs from a de novo assembly. One of these new tools is AutoAnnotate (via BLAST) Auto-Annotate (via BLAST) is similar to Auto-Annotate (local), except instead of using curated sequences on your own…
A game changer for identifying unknown sequences MacVector 18.8 is out and it’s packed with new tools! MacVector 18.8 has tools to help you identify and annotate unknown, unannotated or partially annotated sequences. Batch BLAST allows you to automate BLAST searches for multiple sequences and Auto-Annotate (via BLAST) allows you to automatically annotate multiple sequences.…
MacVector 18.8 requires macOS Big Sur (11) or later, including macOS Tahoe when it is released. It will NOT work on Windows, macOS 10.15 (Catalina) or earlier. MacVector 18.8 is a “Universal Binary”, meaning it will run natively on both Intel and Apple Silicon based Macintosh computers. The changes in MacVector 18.8 are described in the Release Notes .…