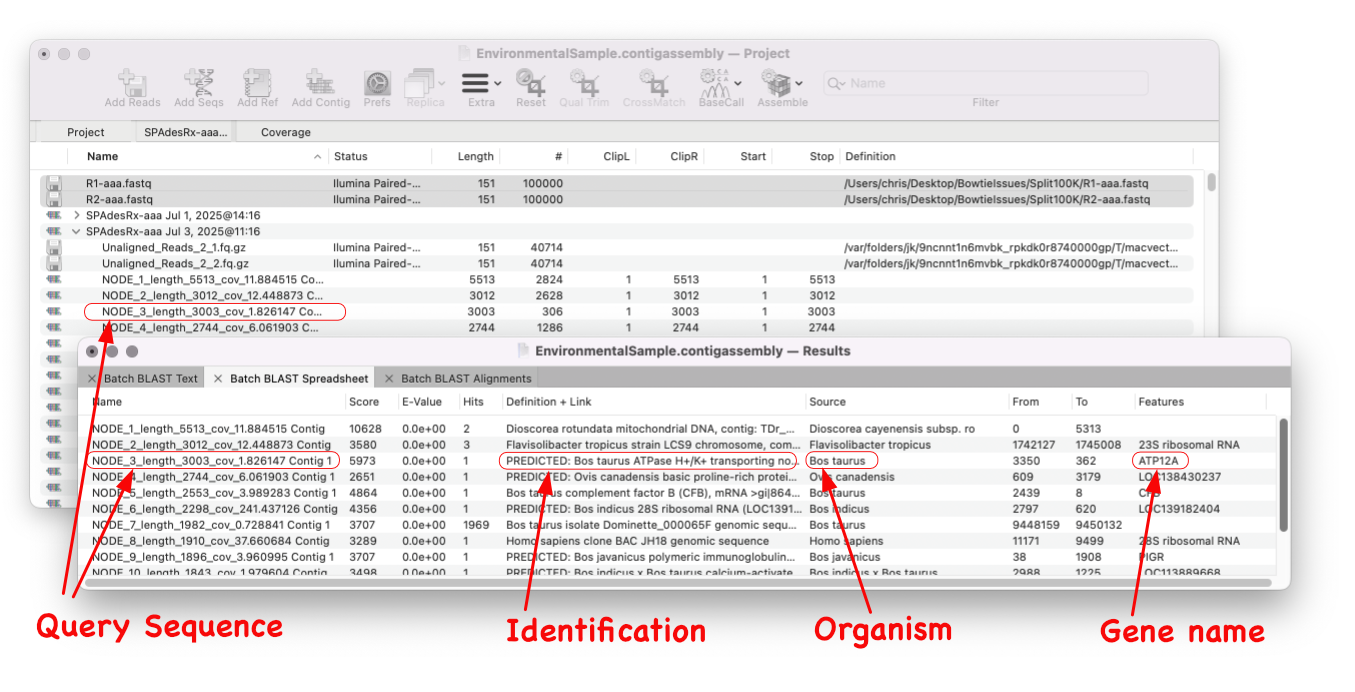
A game changer for identifying unknown sequences MacVector 18.8 is out and it’s packed with new tools! MacVector 18.8 has tools to help you identify and annotate unknown, unannotated or partially annotated sequences. Batch BLAST allows you to automate BLAST searches for multiple sequences and Auto-Annotate (via BLAST) allows you to automatically annotate multiple sequences.…
MacVector 18.8 requires macOS Big Sur (11) or later, including macOS Tahoe when it is released. It will NOT work on Windows, macOS 10.15 (Catalina) or earlier. MacVector 18.8 is a “Universal Binary”, meaning it will run natively on both Intel and Apple Silicon based Macintosh computers. The changes in MacVector 18.8 are described in the Release Notes .…
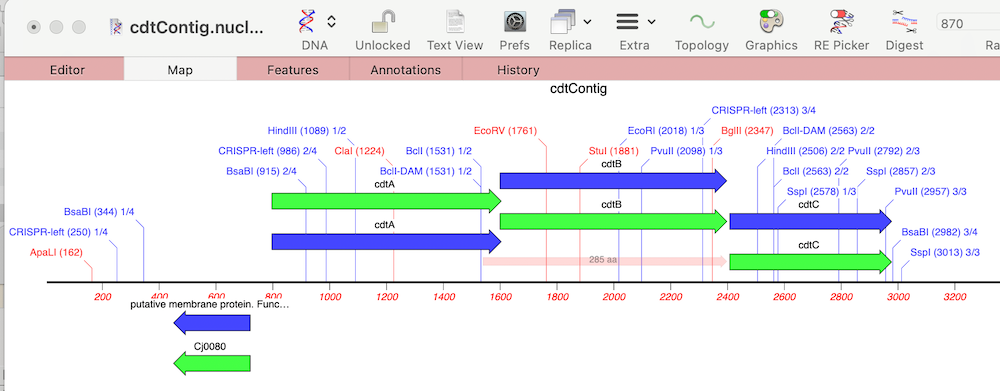
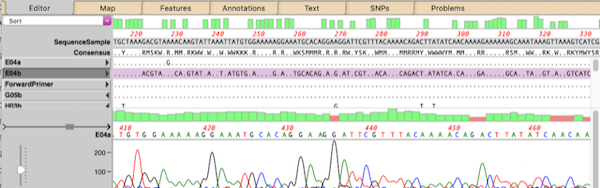
In last week’s tip we showed you how to filter NGS read data to pull out and assemble just those reads that represent a specific gene of interest. Now let’s see how to annotate the single contig we generated and compare that to a reference genome. First, from the Contig Editor, you can save the consensus in MacVector…
Even the latest Macintosh computers loaded with as much RAM as you can afford will still struggle to de novo assemble genomes much over 50 Mbp. But, often that is not required. If you are just interested in a few genes, or a specific region of a chromosome, you can use Align to Folder to filter the…
The majority of MacVector’s analysis tools use a standard workflow: Optimizing results Once you have run the initial analysis then for subsequent analysis you can just repeat the analysis you have just done and further filter it. To do so you need to rerun the analysis tool except now select the Results window (at the…
MacVector Pro now includes Assembler, a powerful sequence assembly plugin that brings sequence assembly directly to your desktop with the same user-friendly interface MacVector users have come to expect. Assembler simplifies the management, assembly, and analysis of all types of sequencing data. Assembler’s extensive toolkit has always been seamlessly integrated into MacVector, but previously required…
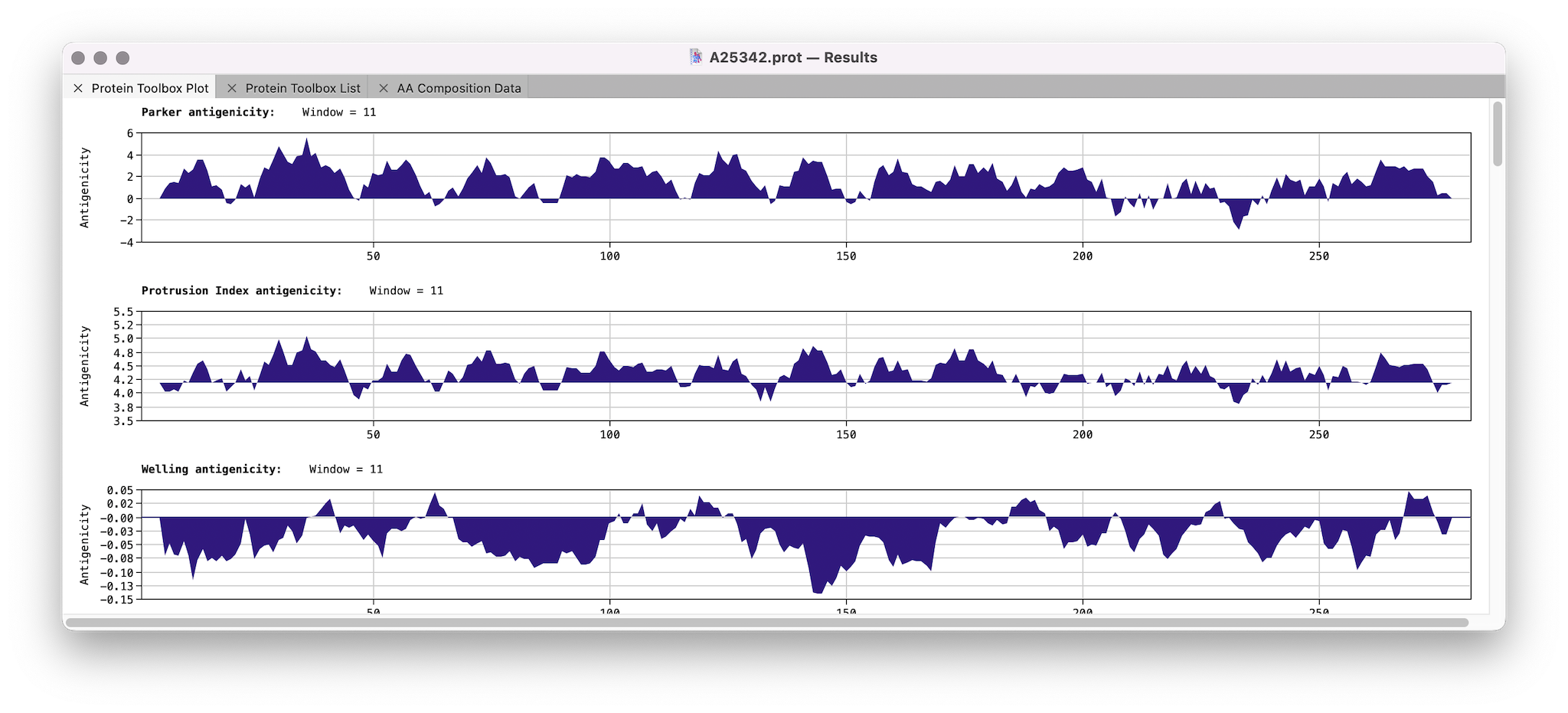
We’ve just released MacVector 18.7.3. This has a few minor bug fixes with calculating amino acid properties of a protein sequence: If you have not yet been prompted to upgrade then go to MACVECTOR | CHECK FOR UPDATES… or download the installer directly. MacVector 18.7.3 is fully supported on macOS High Sierra to macOS Sequoia.
Apple released macOS Sequoia earlier this week (16th September 2024). As usual in the run up to a new macOS release, we have been testing MacVector on development builds of macOS Sequoia. We are happy to report that there are no issues and that MacVector 18.7 is fully supported on macOS Sequoia. If you do come across…
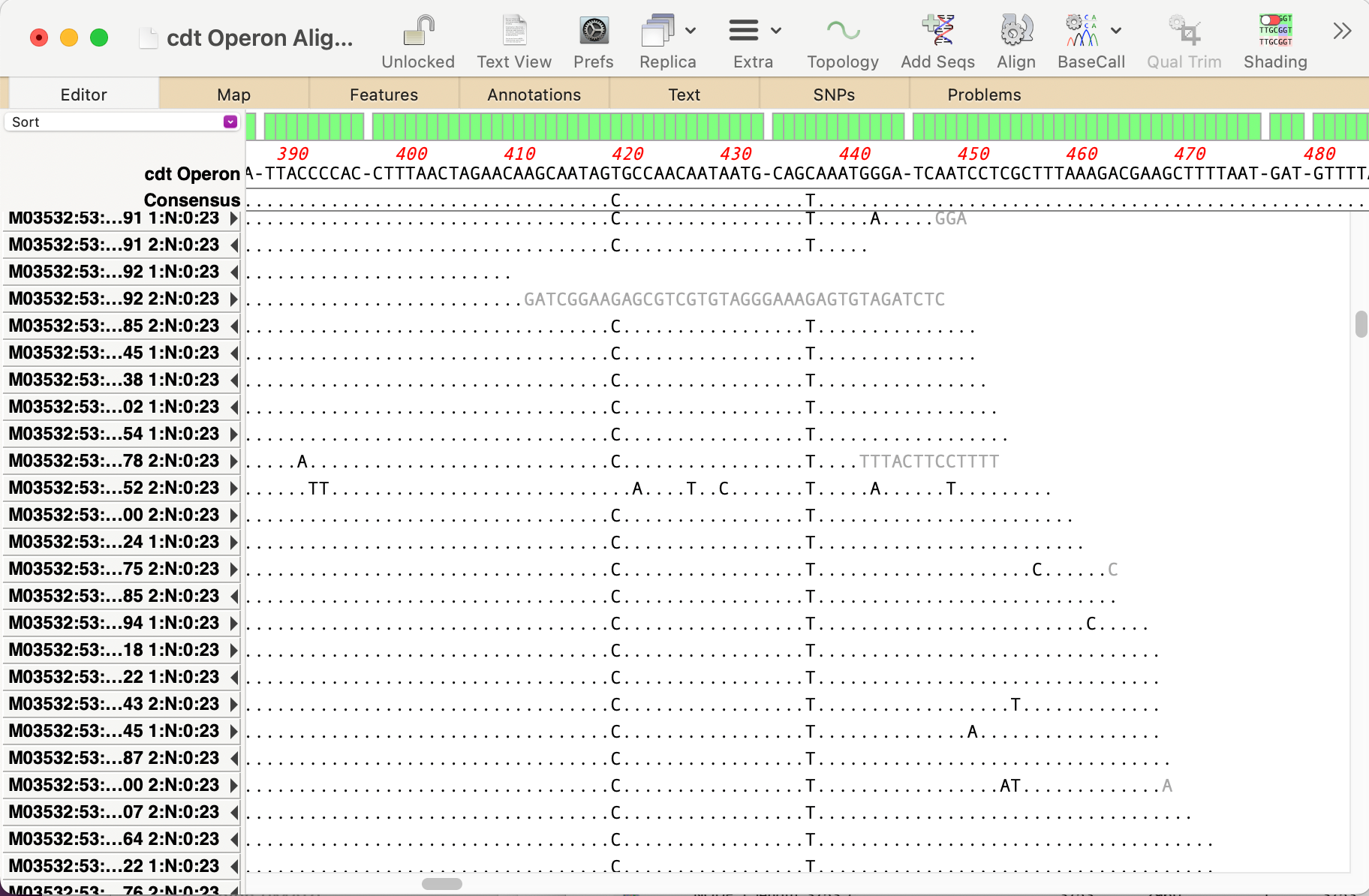
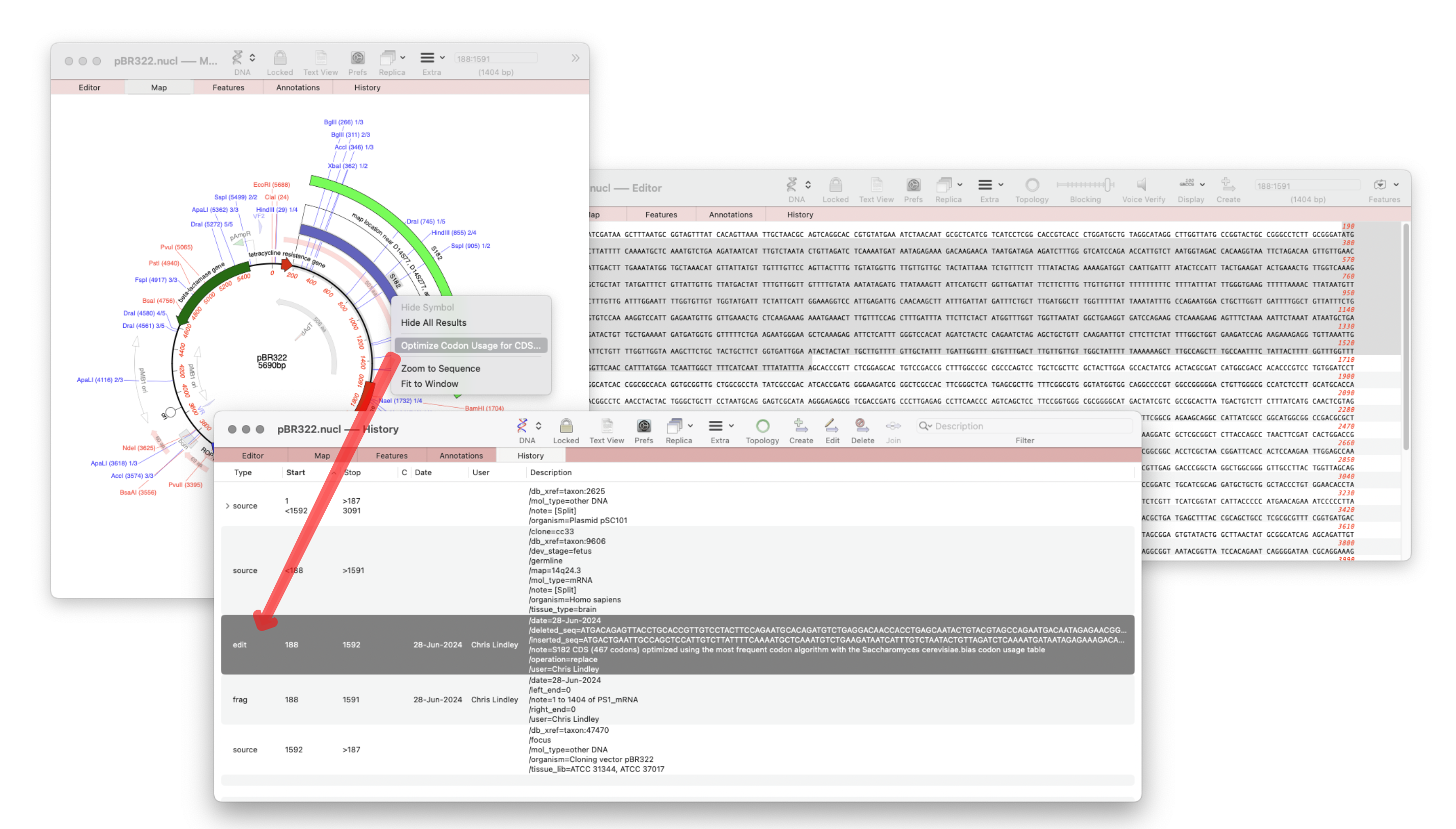
The recently released MacVector 18.7 has a new History tab in the Single sequence editor that shows the editing history of your DNA sequences Since the introduction of MacVector’s Cloning Clipboard, all cloning actions (such as ligating a digested fragment into a vector) create a /FRAG feature that records the source of the ligated fragment, the restriction enzymes used to digest it (and…
One change in MacVector 18.7, that will improve installation on multi user Macs, is that by default MacVector now stores restriction enzyme files in the user’s home folder. Since it’s the user’s home folder, it will always be writeable, even if the user does not have Administrator access to the machine. Additionally for a Mac…