
Tag: assembler
-
MacVectorTip: Trimming trace files by quality
Many of our users are familiar with the ability of Sequencher to semi-automatically trim poor quality sequences from the ends of Sanger ABI reads. Although it is generally not necessary to do this in MacVector because most of the algorithms can automatically handle poor quality data, there are times when it can be beneficial. So…
-
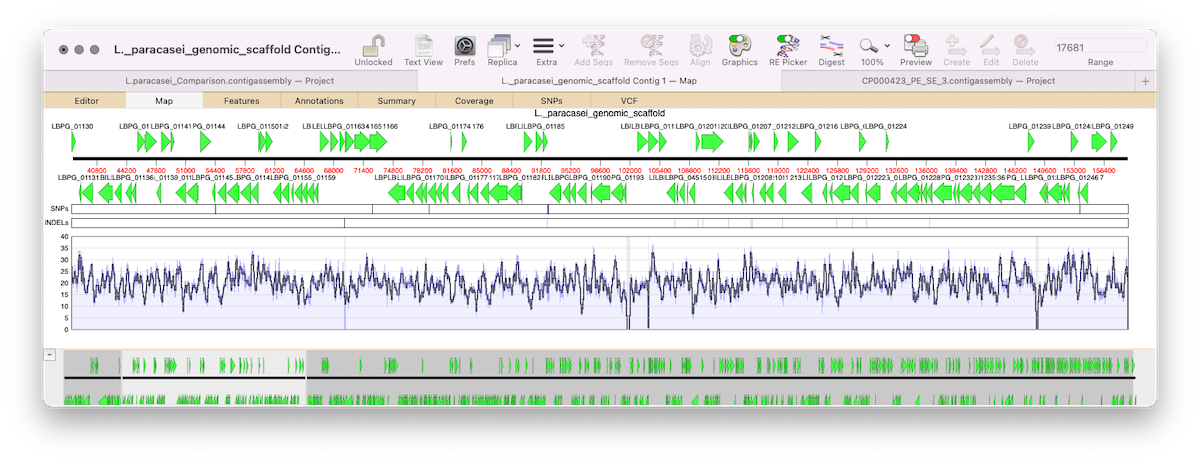
MacVectorTip: correctly flagging PacBio and Oxford Nanopore datasets for assembly by Flye
MacVector 17.5 introduced Flye for assembly of PacBio and Oxford Nanopore reads. Flye joins Phrap, Velvet and SPAdes for de novo sequence assembly using along with Bowtie2 and Align To Reference for reference assembly. Flye is an assembler algorithm tuned to assemble low quality long reads such as those produced by the new generation of…
-
MacVectorTip: quality score visualization in sequence assemblies.
Quality scoring of Assemblies and Align to Reference alignments can be visualized directly on the sequence. Residues can be shaded according to their quality scores. These can be displayed anywhere quality values are available, including de novo and reference assemblies in Assembler and Align to Reference alignments. A Shading toolbar button lets you turn on…
-
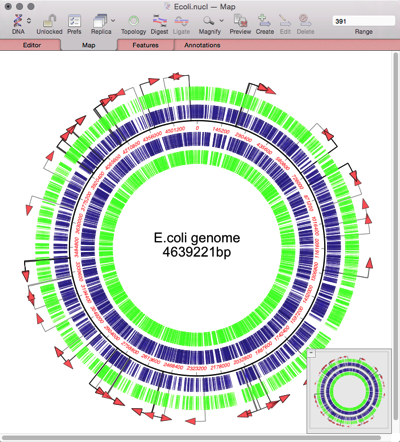
MacVectorTip: Assembling Fungal Genomes using SPAdes
MacVector with Assembler can assemble bacterial genomes in just minutes on quite modest hardware. Currently MacVector has four de novo assembly tools (SPAdes, Velvet, Flye and Phrap). But what of larger genomes? It is currently impractical to run de novo assemblies of Human genomes on a low cost Mac, though RNA-Seq analyses against the human…
-
MacVector 18.2 is out! …and ready for macOS Monterey
Overview We are very pleased to announce that MacVector 18.2 is available to download. MacVector 18.2 is a Universal Binary that runs natively on both Apple Silicon and Intel Macs. It is fully supported on macOS Sierra (10.12) to macOS Monterey (12). New features: Align to Reference Enhancements Context Sensitive Hamburger Menus Where a window has…
-
MacVectorTip: Using the Align to Reference Shading and Trimming toolbar buttons
MacVector’s Align to Reference Editor and the Contig Editor in Assembly Projects have two useful functions for visualizing assemblies. The Shading button turns on background coloring of the residues in the upper pane, based on quality values (these can be from Sanger reads or from NGS reads). The scale ranges from a dark red for…
-
MacVectorTip: Identifying, Selecting and Assembling NGS reads with a variant genotype
When analyzing/assembling/aligning NGS data, there are many scenarios where you might want to separate out the reads representing different genotypes or variant sequences. MacVector makes this very easy. Take a reference sequence and choose Analyze->Align to Reference. Now click the Add Seqs button and select and add your NGS data files. NOTE: if your reference…
-
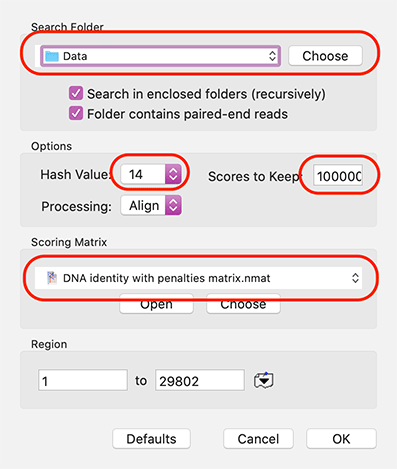
MacVectorTip: Filtering NGS Data to retrieve reads matching a known sequence
So you just got your NGS reads back from that sequencing experiment and, wow, what a HUGE amount of data. Wouldn’t it be easier to handle if you could pare that down to just the gene/plasmid/sequence(s) you are interested in? MacVector to the rescue as it can read and filter fast/q files, even if they…
-
MacVectorTip: Use Bowtie to remove contaminating reads prior to NGS Assembly
MacVector with Assembler can use Velvet and/or SPAdes for fast and memory efficient de novo NGS assembly of modest sized genomes (typically up to 40 Mbp or so) even on a laptop. One common task is to assemble NGS data from BAC clones.However, one problem that often arises is that the BAC DNA preparations may…
-
MacVectorTip: Context-sensitive Menus in MacVector
Although Apple are well known (notorious?) for always providing mice with only a single obvious button, in reality the Mac interface from early versions of MacOS all the way to macOS Big Sur, plus many Mac apps, have always used right click menus (or more accurately “context sensitive menus”) to provide extra functionality. MacVector is…