
Author: Chris
-
Working from home: An overview of assembling sequence data with MacVector and Assembler
Working from home Here’s a series of blog posts on the wide range of functionality in MacVector that you may never have used before. This is an overview of the many different sequence assembly tools within MacVector. The MacVector team used these tools to mine existing sequencing archives to assemble a new Pangolin SARS-CoV–2 genome. To…
-

Accessing video tutorials of common workflows inside MacVector
During the Covid-19 pandemic we want to ensure that you have access to the MacVector license that you would use in the lab, if you are working from home. If you use MacVector, even an older version, and are having trouble activating it (or installing it) at home email MacVector Support and we will help. If you…
-
Working from home – Getting your sequence into MacVector
During the Covid-19 pandemic we want to ensure that you have access to the MacVector license that you would use in the lab, if you are working from home. If you use MacVector, even an older version, and are having trouble activating it (or installing it) at home email MacVector Support and we will help. If you…
-
Working from home with MacVector during the COVID-19 pandemic
A lot of MacVector users are now at home getting used to a new way of working. The MacVector team are distributed throughout the US and Europe and we are used to remote working. However, for those new to working from home, it’s a LOT different to working back in the lab with your colleagues!…
-
Primer validation with MacVector: Primer3, Covid19 and primer design
The CDC recently published diagnostic real-time primers for identification of SARS-CoV–2 in any person suspected of having COVID–19. Unfortunately as pointed out on the Biome Informatics blog these primers have issues that should have easily been detected had the primers been tested using a good quality primer testing tool (the linked blog post uses Primer3).…
-
MacVector Training workshop at The Crick: Tuesday 17th March 2020
Unfortunately due to the Covid19 outbreak this workshop is now cancelled. Keep safe and healthy everybody. The workshop, previously cancelled last year, is now rescheduled: Room: HR Training Room 01.2162. Floor: 1Date: 17th March 2020 – from 9:30 – 11:30 Chris Lindley of MacVector, Inc. will be giving a training workshop for both novice and…
-
Make more of your alignments with MacVector 17.5
Our latest release MacVector 17.5 gives you new tools to make the most of your alignments. It displays shared domains in protein alignments to visualize the relationships between aligned proteins. It introduces Flye for de novo assembly of PacBio and Oxford Nanopore long reads and a slew of enhancements to the Contig and Align to…
-
Importing BAM files into an Assembly Project
You can import BAM files, containing reads mapped against a reference sequence, into a MacVector Assembly Project. As well as the BAM file(s) you will also need the original reference sequence the reads were mapped against. FASTA is fine, but an annotated reference is better for visualisation. The tool needed is called ADD CONTIG. This…
-
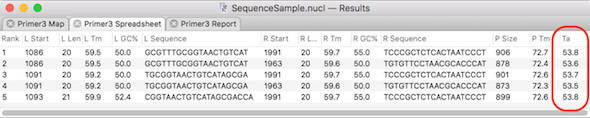
Calculating the optimal PCR annealing temperature
MacVector has several tools to help with primer design and testing. The Analyze | Primer Design/Test (Pairs) function uses the popular Primer3 algorithm to find suitable pairs of primers to amplify specified segments of DNA. You can also enter pairs of pre-designed primers and test their suitability for use in PCR. In both cases, the…
-
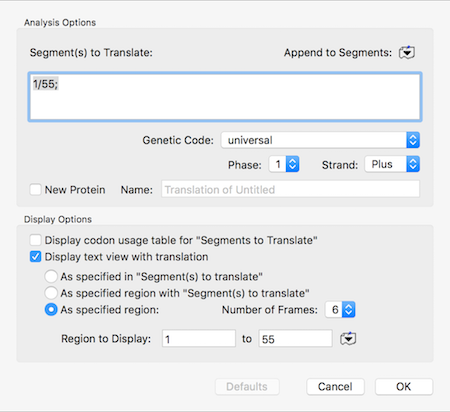
How to copy a specific short amino acid translation of a sequence
There can be times when you are messing about with open reading frames, inserting residues to change frames to try to get the perfect CDS fusion. The MacVector single sequence Editor will show those (click and hold on the “Display” toolbar button) but if you select and copy, only the DNA sequence (with any overlapping…