What's New in MacVector 16?
Overview
MacVector 16 adds an automatic search for missing features whenever you open a DNA sequence. This complements the existing searches for ORFs and Restriction Enzymes. There is also a new Feature Editor that lets you change the underlying "GenBank" data as well as the visual symbol appearance in the same dialog. Assembler has a new de novo assembler algorithm (SPAdes) as well as improvements to the handling and analysis of many different types of NGS data. You can view the release notes here.
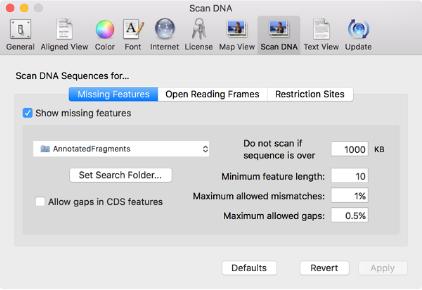
Scan For Missing Features
Whenever you open a DNA sequence, MacVector now scans it to identify common features that are not already annotated on the sequence. It shows these in faded colors in the Map tab so that you can see what features are missing from your sequence. By default, MacVector uses the collection of features in the /MacVector/Common Vectors/Annotated Fragments/ folder, but you can use any folder you like, configured using the new MacVector | Preferences | Scan DNA preference settings.
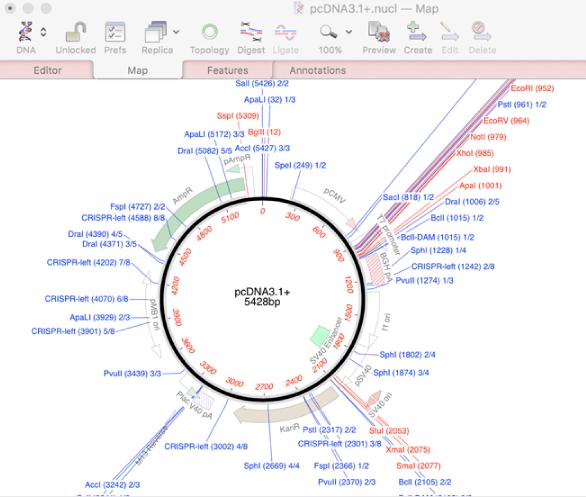
If you open a typical unannotated vector sequence, you will get a display something like this;
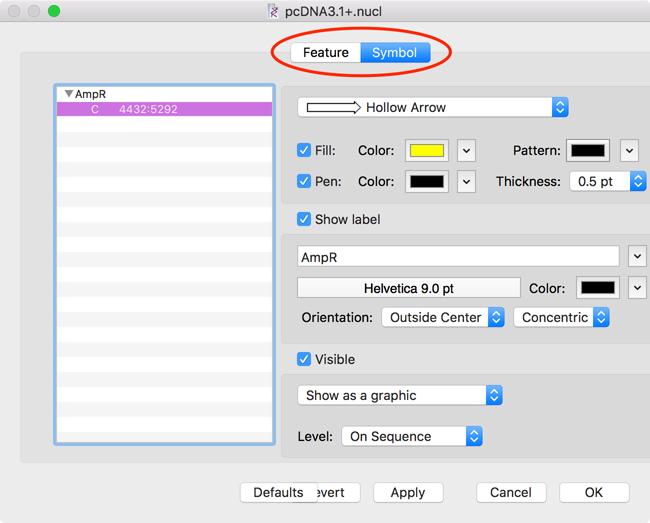
Combined Feature/Symbol Editor
If you double-click on an individual feature in the Map tab, it now brings up a dialog with two tabs – one for the Feature Editor and one for the Symbol Editor. This lets you edit both the basic Genbank-style feature information and the graphical symbol appearance in a single dialog. Note: this is only displayed when you are editing a single individual feature. If you want to change the appearance of multiple features at the same time, you will get the normal Symbol Editor.
Assembler Improvements
We have added the popular SPAdes algorithm to Assembler for MacVector 16.0. This is a high-performance assembler that uses very little RAM, allowing it to run on relatively modest machines, though we do recommend 16GB+ of RAM for optimal performance. It is a little slower than our existing velvet algorithm, but does a better job of resolving repeats at the ends of contigs, so you generally find it produces longer contigs with the same input data. One drawback of SPAdes is that it does not generate alignments, only contig consensus sequences, so we have added an option to generate alignments within MacVector using bowtie. This does increase processing time.
You can now use interleaved paired read files for all NGS assembly algorithms (velvet, bowtie and SPAdes). MacVector will automatically identify them where possible.
MacVector now directly supports compressed fasta and fastq formatted files (.gzip/.gz) and will submit those directly to assembly algorithms. This dramatically reduces the amount of temporary disk space required when assembling large datasets.
There are now more options to control the type of read that you are using (unpaired, paired-end, mate pair, HQ mate pair, NxSeq long mate pair), or the source of the reads (Illumina, IonTorrent, PacBio, Nanopore etc). Double-click on a read after importing into a project to set these parameters. SPAdes in particular will optimize assembly based on these settings.
There is now a right-click option to circularize contigs in the Contig Editor. If there are direct repeats at the ends of a contig, you can right-click and the item will show you the length of the repeat. If you select the item, a new circular sequence will be created from the consensus.
Miscellaneous Changes
In the single nucleic acid sequence Editor view, when you hover the pointer over a residue, the tooltip now displays the nucleic acid residue number under the pointer, and also the amino acid residue number of any annotated CDS feature at that position.
The Align To Reference assembly algorithm has been improved to better handle reads crossing the circular origin of a reference sequence. It is also now significantly faster when aligning longer reads with multiple regions of mismatches.
Multiple Sequence Alignments now have an option to “unalign” selected sequences, or the entire alignment – it’s an option on the Align toolbar button.
By default, MacVector now uses a smaller arrow for ORF results, with a pale red color for matches on the plus strand and pale grey for matches on the minus strand to distinguish them from annotated features.
MacVector 16 has had yet more functions added to the AppleScript directory. To see what functions are available, open the apple-supplied Script Editor, choose File | Open Dictionary and select the MacVector.app application. For examples of using AppleScript to drive MacVector, see sample files in the /Applications/MacVector/AppleScripts/ folder.
|

