What's New in MacVector 17.5?
Overview
MacVector 17 has a cool new domain-outlining facility for multiple sequence alignments, letting you more easily visualize the relationships between features in aligned sequences. There is a new algorithm (Flye) for de novo assembly of PacBio and Oxford Nanopore long reads and a slew of enhancements to the Contig and Align to Reference Editors, including coloring based on quality values and editing status. As ever there are a slew of minor enhancements, bug fixes and changes to better support the latest releases of macOS.
Outlining Shared Domains in Aligned Sequences
Multiple Sequence Alignments now retain feature information from their individual input sequences and can use this information to outline shared domains in the aligned sequences. To use this feature, first individually annotate the sequences you want to align, make sure the domains/features you are interested in are visible and set the Fill color to the color you would like to see in the alignment. Then add the sequences to a multiple sequence alignment document and align in the usual way (or, keep the single sequence documents open and choose Analyze | Align Multiple Sequences Using...). Then click on the Mode toolbar button (shown below) and select Show Features
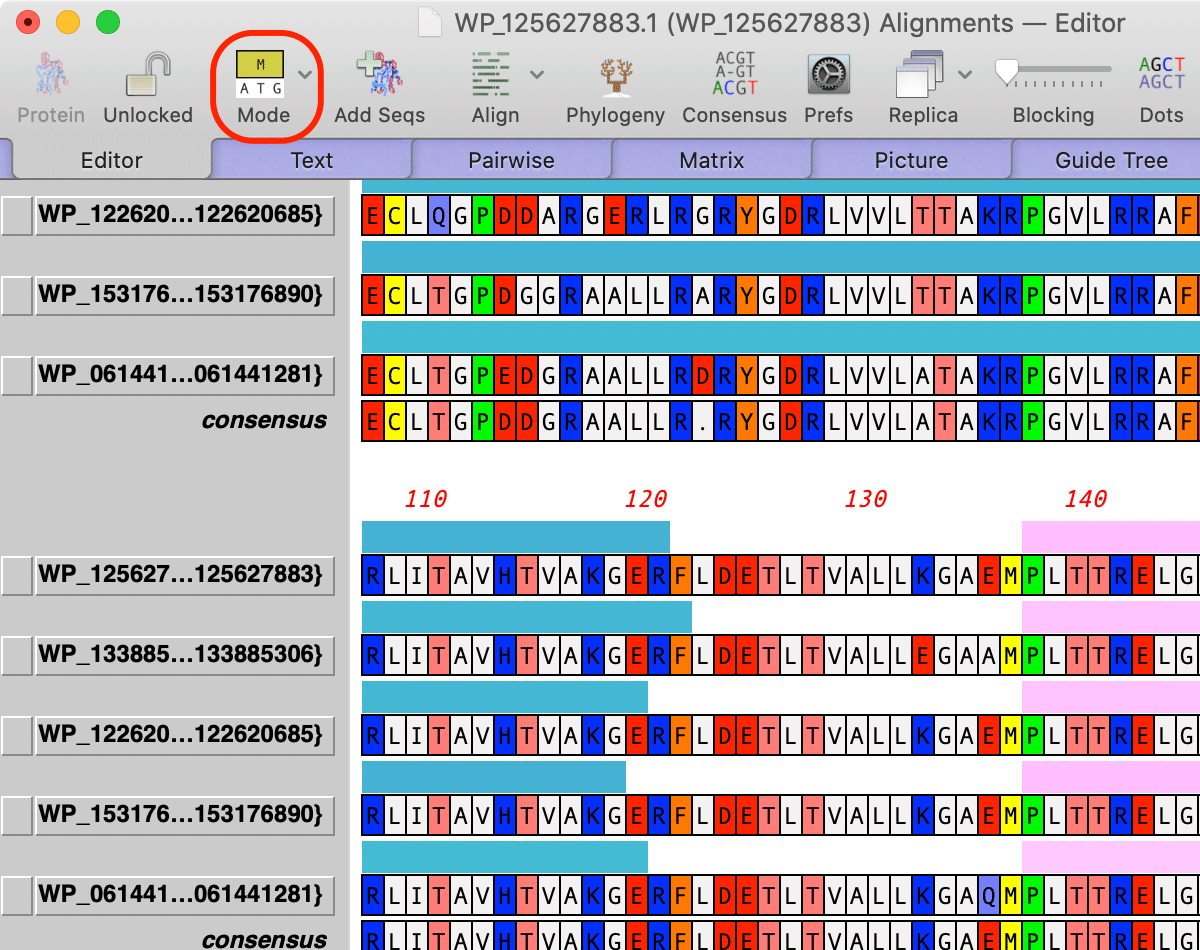
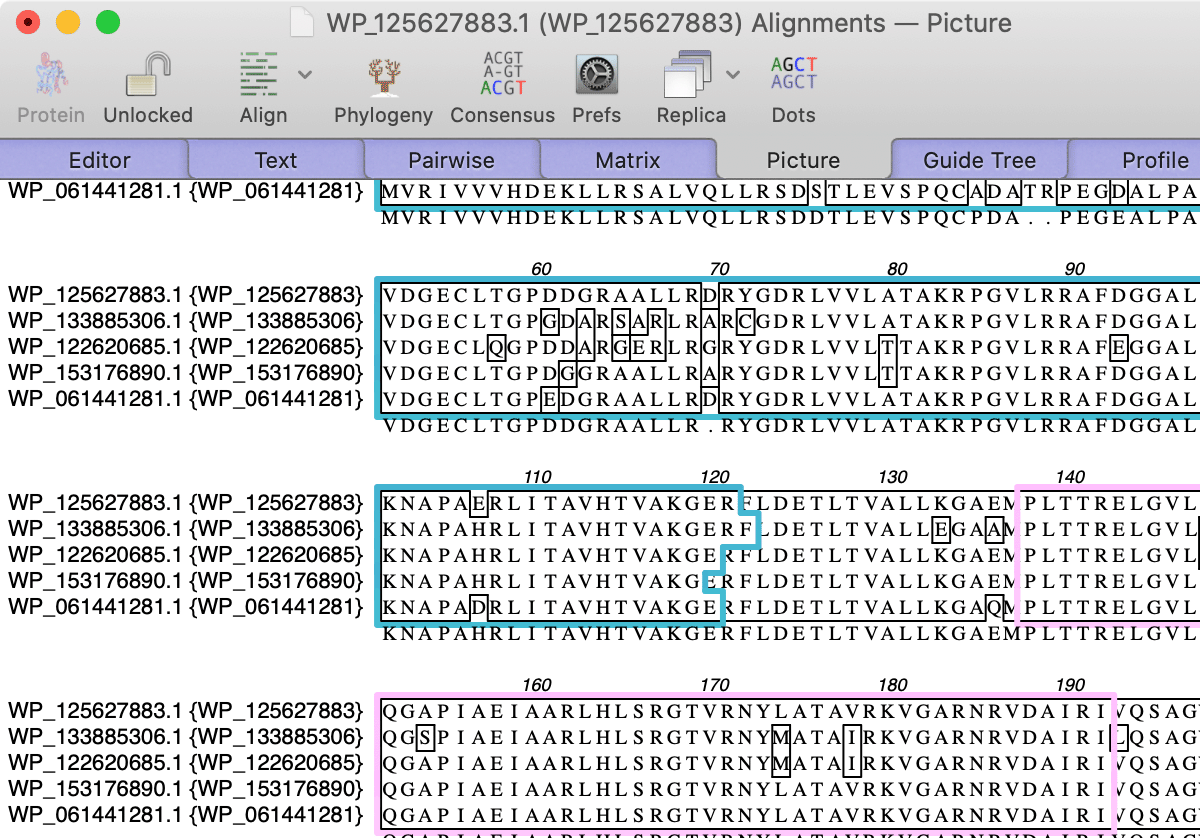
This turns on a simple feature display mode in the Editor tab where you can see the extent and color of the features. When you switch to the Picture tab, you will see colored outlines around the shared domains;
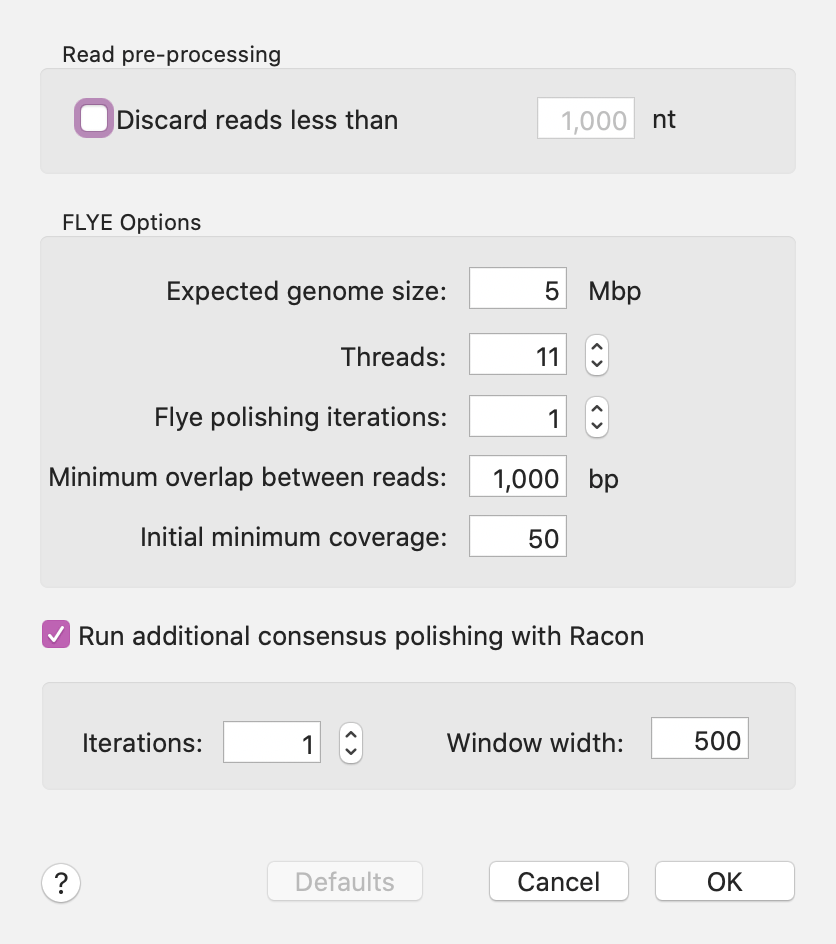
de novo Assembly of PacBio and Oxford Nanopore reads with Flye
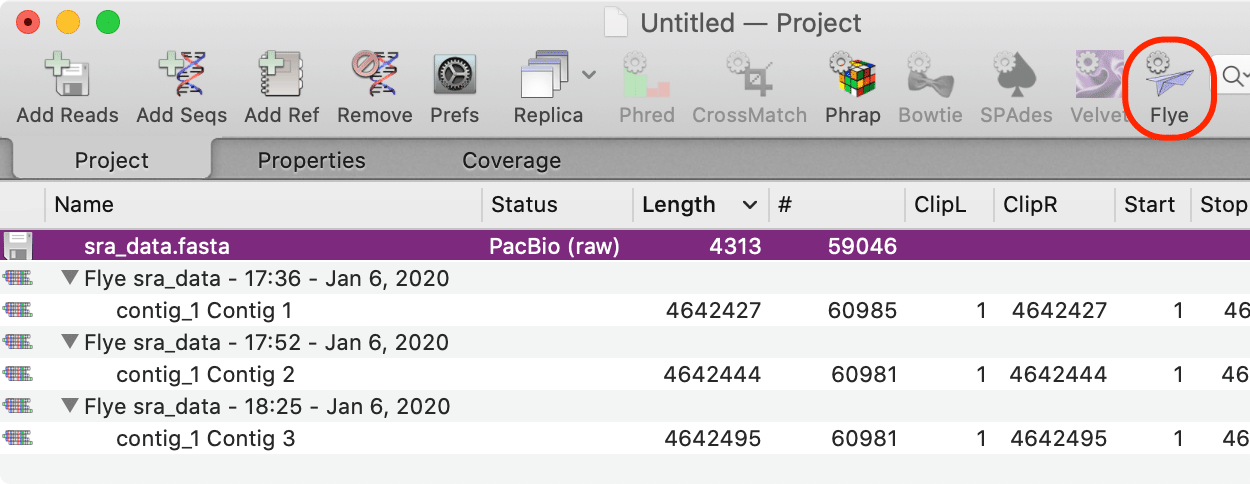
Flye is an assembler algorithm tuned to assemble poor quality long reads such as those produced by PacBio and Oxford Nanopore sequencers. Because these reads tend to be very error prone, MacVector 17.5 also includes an optional polishing step using Racon. With typical bacterial genome assemblies it is fairly common to be able to assemble reads into a single full-length genome contig, such as with this set of reads from a strain of E. coli;
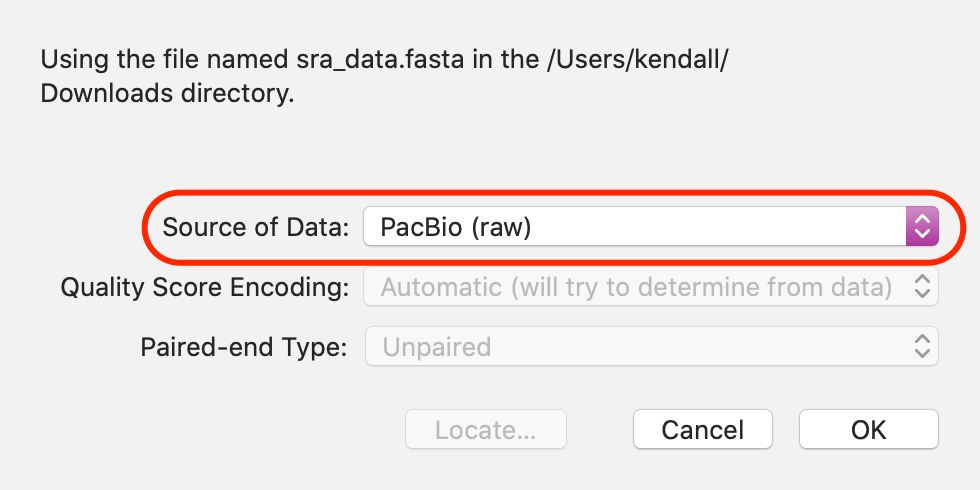
Note that it is important to tell MacVector what type of reads you are assembling - this is easily done by double-clicking on the Status item after importing the reads via the Add Reads toolbar button.
The three jobs above are the result of varying Flye parameters. However, all three resulted in a single contig approximately matching the size of the E. coli genome - the differences in length reflect different "polishing" strategies with the last one having one round of internal polishing by Flye and one additional round of polishing by Racon.
As one of the last steps in Flye assembly, MacVector aligns the input reads against each contig consensus using minimap2 so that you can view the alignments in the Contig Editor. Because of the noisy nature of Long Read sequence data, MacVector removes additional erroneous inserted residues in each read prior to display to clean up the visual alignments.
Contig and Align to Reference Editor Enhancements
There have been a number of enhancements to these editors, primarily to aid in visualizing edits and quality values and to "clean up" the visual appearance of alignments.
Residue Background Colored by Quality
A Shading toolbar button lets you turn on coloring based on the quality value assigned to each residue;
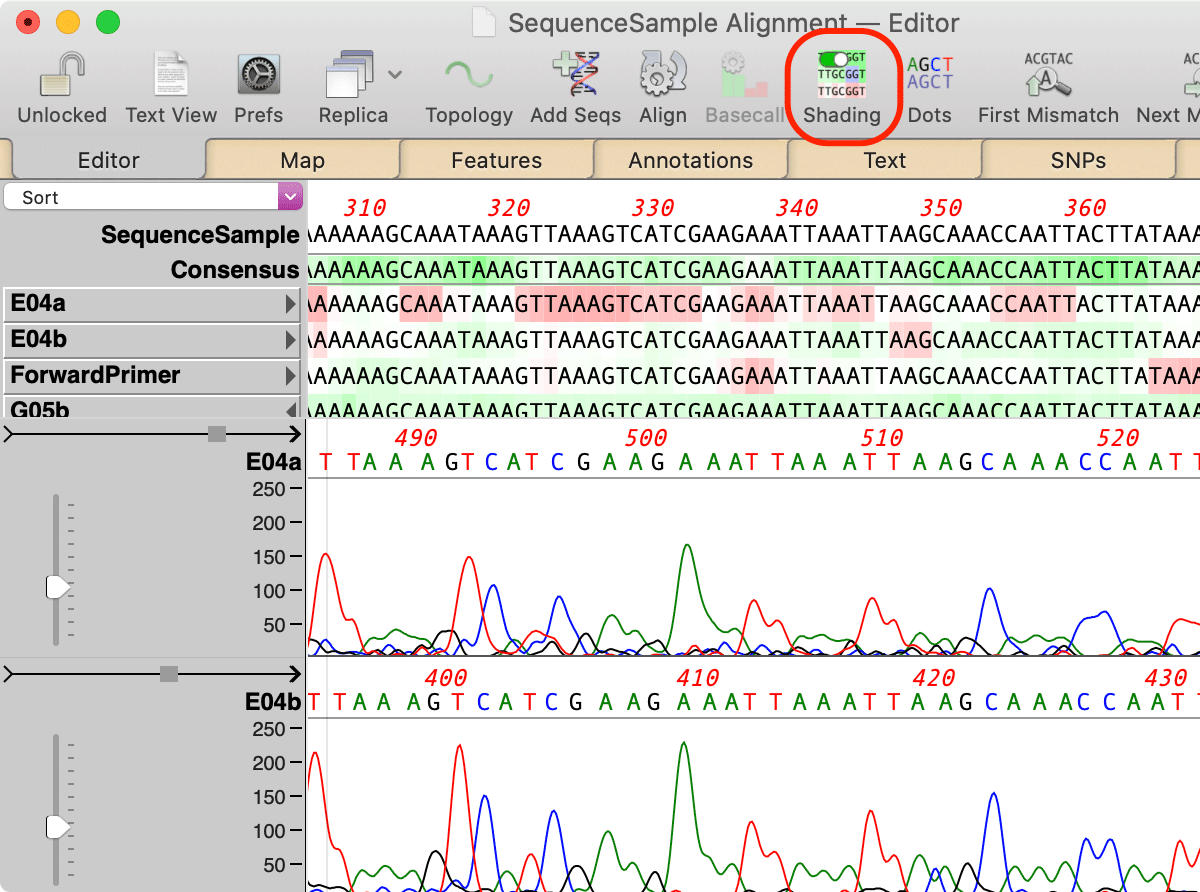
The intensity of the colors indicates the phred-based quality value of each residue. For individual reads, this ranges from 0 (deep red) through 20 (white) to 40 or above (deep green). The consensus scale is doubled and ranges from 0 (deep red) through 40 (white) to 80 or above (deep green). Gaps are always shown with a white background. As with earlier versions of MacVector, you can "mouse-over" a residue to view the numerical information in a tooltip.
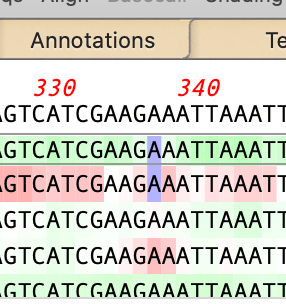
Edited Residues Have a Blue Background
Edited residues are always given a phred quality value of 99 - these residues are given a blue background;
Base Calling with Phred
You can now directly run phred on Sanger sequencing trace files in the Align to Reference Editor by clicking on the Basecall toolbar item with the appropriate sequences selected;
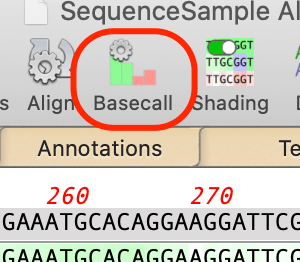
Note that you must do this before aligning the sequences. You can always "reset" (i.e. un-align) and sequence by selecting its name and choosing Reset (un-align) Selected Reads from the right-click (or <ctrl>-click) context-sensitive menu.
Editing Enhancements
There are some new context-sensitive menu items in the Align to Reference Editor tab
Delete Clipped Residues - deletes any greyed-out ("clipped" or "trimmed") residues. While these are ignored by the consensus calculation, some users prefer to delete them for a cleaner looking alignment.
Close Gaps by Deleting Residues - you'll often see gaps in the consensus where one or more reads has an additional erroneous inserted residue. This menu item removes the extra residues from the read, cleaning up the visual appearance of the alignment.
You can now "nudge" reads in the Align to Reference Editor tab. Select the name of the sequence you want to nudge and use the left/right arrow keys to move it around. If you have problematic alignments where you need to physically insert residues or gaps, hold down the <option> key when typing to insert the residue or gap before the currently selected residue, then use the nudge function to get the alignment right. The consensus updates in real time.
Miscellaneous Enhancements
There have been a large number of minor enhancements. Some, such as reworking code behind the scenes to replace deprecated Apple functions and refactoring code for better stability and performance help ensure that MacVector will continue to work on upcoming releases of macOS and take advantage of improved hardware. There have also been improvements to Dark Mode support in many area and much better handling of the labels in crowded Map views.
|

