What's New in MacVector 18.5?
Overview
MacVector 18.5 adds a number of new functions, particularly regarding the identification of heterozygotes and mixed residues in ABI and Beckman chromatogram based Sanger sequencing data.
MacVector 18.5 is a Universal Binary, meaning that it runs natively on both Intel and Apple Silicon ("M1" or "M2") Macintosh computers. It requires a minimum of macOS 10.12 (macOS Sierra) and is certified on macOS Monterey.
Heterozygote Analysis
There is a new Heterozygote Analysis item in the Analyze menu. It is active whenever an individual Trace ("chromatogram") window is frontmost, or when Align to Reference or Assembly Project windows containing trace/chromatogram data are frontmost. This invokes a custom algorithm that operates on the chromatogram data of an individual trace, or of all traces in an Align to Reference or Assembly Project window. The parameters are fairly basic;
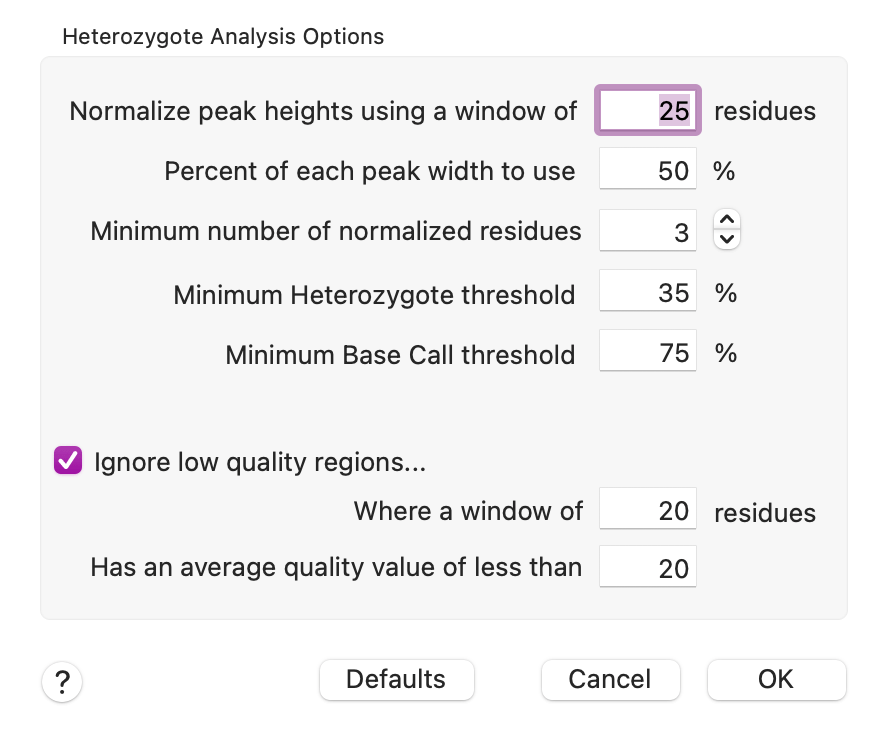
The algorithm understands that the quality and signal strength of chromatograms can vary considerably between tracks (e.g. A's might be much stronger than G's in an individual run) and also across the length of a run. So you can control the width of the window used to normalize the data, along with the minimum number of residues for each track. So a value of "3" means the algorithm will extend the window until at least 3 peaks have been found for each track. You can also control how much of each peak is used for the calculation - using less than 100% helps to reduce cross-contamination from adjacent peaks. The algorithm also separately adjusts the "bleed" from adjacent peaks to better isolate the signals for each individual peak. Finally, the Heterozygote threshold value controls how much signal is required before calling a heterozygote. There is also an option to skip regions of low quality in the trace data.
After running, the results are displayed in a standard text window with hyperlinks that jump to the heterozygote residue in the sequence. For single trace files and for Align to Reference windows, this simply scrolls to the appropriate location. For Assembly Project windows, the corresponding trace window gets opened.
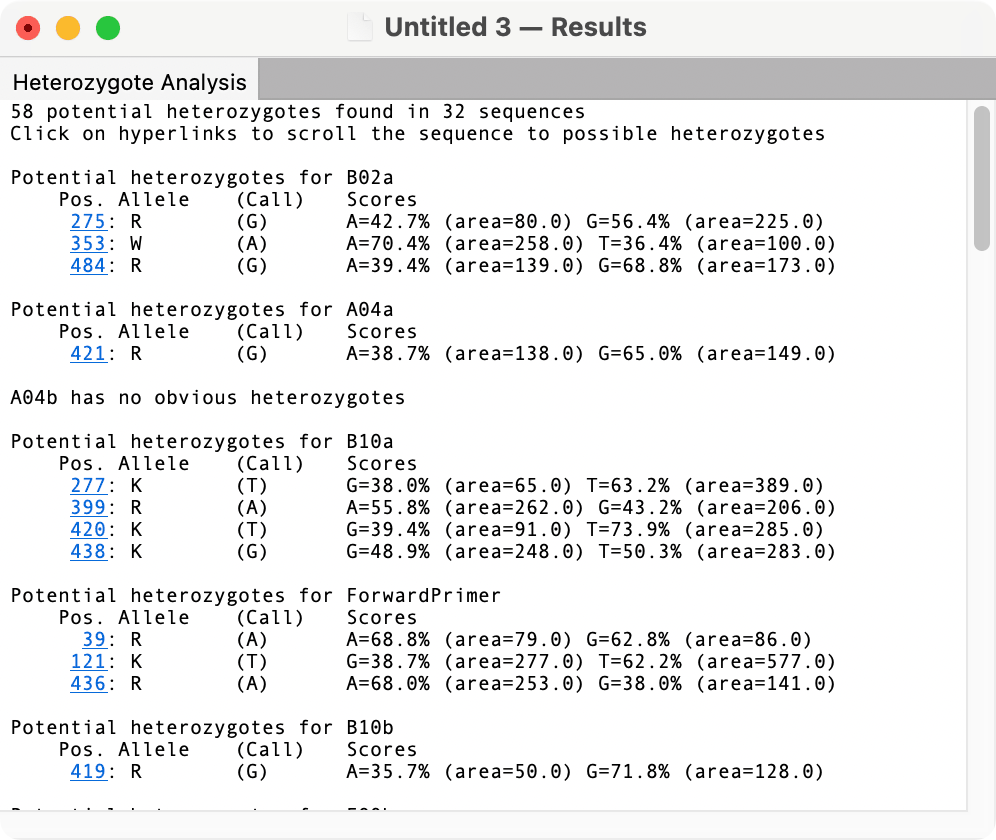
Heterozygote Basecall
There is an additional variant of the heterozygote analysis that automatically changes the identified heterozygous residues to the corresponding IUPAC ambiguity code. The algorithm and result windows are identical to the Heterozygote Analysis function, but there is the added option of applying the changes to the target sequences. Particularly with Assembly Projects, this is a great way of rapidly re-calling potentially thousands of Sanger sequencing files with possible heterozygotes.
Miscellaneous Enhancements and Bug Fixes
There have been significant improvements to MacVector's ability to read Sequencher Project (.SPF) files. Additional enhancements will be available shortly.
In the Align to Reference Editor, the 3/6 frame translations now extend beyond the ends of the reference sequence if the aligned consensus sequence is longer than the reference.
Flye has been updated to version 2.9.1 and now uses Python 3.9 for improved performance.
Various sheets and dialogs have been cleaned up to allow for the different fonts and spacing in macOS Ventura
Enable Mixed Case Entry is now enabled as expected.
Auto-annotation jobs that fail are now cleaned up correctly so that they can be re-run without needing to exit MacVector.
A Multiple Sequence Alignment error where ClustalW would sometimes fail to run has been resolved.
A rare MSA crash bug has been fixed.
The Primer3 implementation now supports the primer_max_end_gc variable.
Improved the import of subsequence data to handle very short subsequences.
|

