What's New in MacVector 17?
Overview
MacVector 17 is one of the most feature-packed updates that we have ever released. Highlights include a new interactive Restriction Enzyme Picker, a new and unique genome comparison tool and a highly interactive tool to help you design and document Gibson and LIC cloning experiments. In addition, MacVector 17 has support for the macOS Mojave "Dark Mode", a new interface to help identify genome sequencing errors, automatic searching and display of primer binding sites, addition of Job Objects to assembly projects and numerous performance improvements. Last, but certainly not least, there's a new How Do I? menu that has links to help pages and videos showing you how to perform a wide variety of common tasks with MacVector. You can view the release notes here.
Dark Mode Support
The most striking visual change to MacVector is that many of the colors and icons change to improve readability in macOS Mojave "Dark Mode";
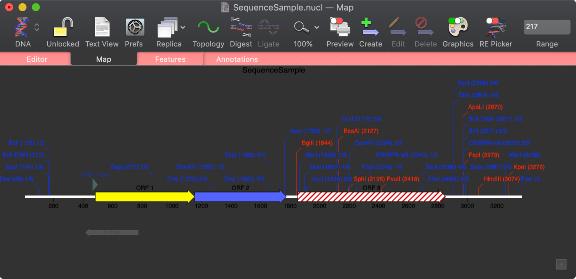
You can read much more about Dark Mode support on this page.
Restriction Enzyme Picker
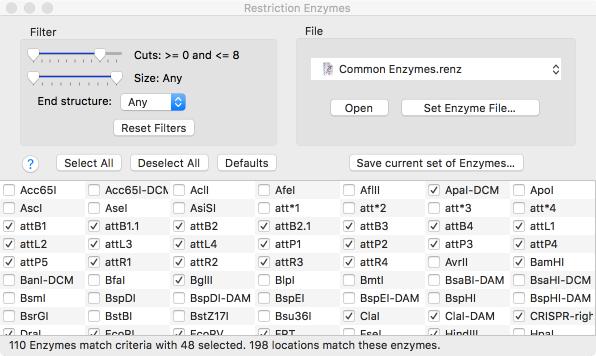
A new floating RE Picker window lets you quickly filter the restriction enzyme sites shown in the Map tab. The window shows all of the enzymes that cut the sequence in the frontmost window, even if they are not currently selected for display. Sliders and popup menus let you select the range of cuts, the type of sticky ends produced etc. You can even save any set of enzymes for use in additional analysis.
Not interested in having the RE Picker visible all the time? All Map tabs now have a simple toolbar button to turn it (and the floating graphics palette) on and off;
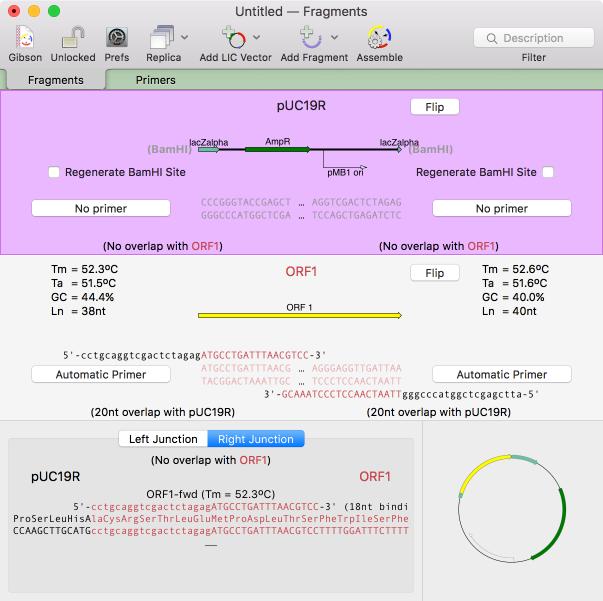
Gibson/LIC Assembly Projects
There is a brand new primary document type aimed at designing and documenting restriction enzyme free assemblies using the popular Gibson and Ligase Independent Cloning techniques. You can use this to help you design PCR primers suitable for the assemblies, or to simply assemble PCR fragments you have already created. The highly interactive interface lets you see the actual CDS translations at the fragment junctions and lets you insert additional residues to adjust the frames appropriately. You can use your own custom primers or let MacVector design suitable primers for you. Adding vectors and fragments to the window could not be easier - you can drag and drop, add from file, or copy and paste. All features in the source fragments are preserved so that you can generate a fully annotated assembled circular product. Read more...
Scan For Primers
A new Scan for.. Missing Primers joins the existing Scan for... tools that automatically display restriction sites, missing common features and putative open reading frames on your DNA sequences. You can control this using the MacVector | Preferences | Scan DNA preference settings.
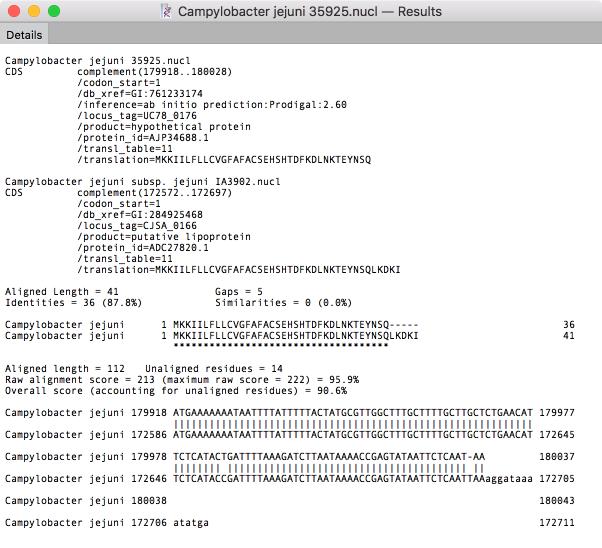
Genome Comparison
In recent years that has been an explosion of whole-genome sequencing projects. One common question coming out of this has been to ask “Exactly what are the genetic differences between my sequenced organism and another related strain?” MacVector to the rescue! A new Analyze | Compare Genomes By Feature… function lets you see the differences between two annotated genomes in fine detail. The algorithm takes every annotated feature from the source genome and looks for the presence of that feature in the comparison genome based on sequence similarity. CDS features are even translated so that the predicted amino acid sequences are compared. The results are then tabulated to show identical, closely related, and weakly related features in separate tabs, with additional tabs for features that are completely missing and a “details” tab that shows the low-level alignment details for any matching pair of features. Hot-links in the result tabs let you quickly scroll the parent sequences to any individual feature of interest.
Assembler Improvements
You can now run multiple jobs in an assembly project and each job is encapsulated in its own job object in the Assembly Project window. This gives you much greater flexibility in organizing your assembly tasks and each job is given an intuitive name.
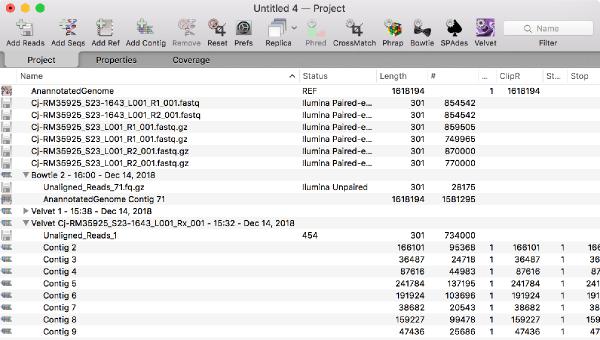
Coverage Tab
Assembly projects now have a Coverage report that allows you to select a reference sequence and directly compare multiple reference assemblies. You can compare different sequencing datasets assembled against the same reference sequence with expression level comparison. Now that you can store multiple assembly jobs in a single Assembly Project, then you can also directly compare multiple runs of the same dataset against a single reference sequence to compare which is the best assembly.
Assembly Problems
Align to Reference now has a Problems tab that helps you identify mis-assembled genomic sequences based on excessive mismatches, discontinuous reads and other common problems. The primary aim of this tab is to help you identify potential assembly issues with genomic sequencing projects. You can use Align to Reference to assemble several million reads against a typical small genome (practically, up to ~10 Mbp or so) and the tab will report those regions that are most likely to be misassembled.
Use of APIKeys with Entrez and BLAST
The NCBI have introduced a new system for accessing the Entrez database over the internet. They now throttle general accesses to the Entrez server to no more than 3 calls per second per IP address. This might seem a lot, but if you want to download a list of 100 sequences, this can really slow things down, especially if you are sharing a public IP address behind a company or university firewall and others are using MacVector. They have introduced a new “APIKey” concept – you can request one of these from the NCBI for your own personal use and that increases the permitted rate to 10 calls per second and is independent of the IP address. Thus, you can share a public IP address with colleagues and still each have the full 10 calls per second access.
Here’s what the NCBI have to say about APIKeys.
Once you get one, you can tell MacVector to use it using the MacVector | Preferences | Internet tab;
How Do I?
There’s a brand new How Do I? menu that shows common workflows with simple step by step guides and short videos.
Every tool dialog now has direct access to a simple video/tutorial on how to use it.
Miscellaneous Changes
You can now copy and paste features between sequence documents. Simply select the feature in the Features tab of one sequence, Edit | Copy, then switch to another sequence Features tab and choose Edit | Paste.
You can now select a short sequence in a Read in the Align To Reference Editor tab, right-click to bring up a context sensitive menu, and then select all of the other aligned reads that contain that a sequence. It’s a great way of selecting all the reads with a specific SNP(s). Combine it with selecting the pairs for each read with NGS data and saving the selected reads and you have a very powerful way of analyzing SNPs or resolving repeat sequences in genomic assemblies.
Some of the nomenclature and interactions in the Primer3 dialog box have been cleaned up to simplify its use.
You can now add sequences to the Align To Reference window from multi-sequence GenBank files.
GenBank multi-sequence files now open by default as individual sequences.
When results are generated for the Bowtie Coverage tab, the name of each feature is now displayed exactly as per the label used in the Map tab allowing you more control over exactly how the text appears.
There is now an option in the Database | Auto-annotate Sequence… interface to automatically “fix” CDS features. This will change the stop location if the match creates a longer or shorter open reading frame and will also change the contents of any /translation= qualifier to reflect the new translated coding sequence.
The Align to Reference algorithm is now multi-threaded. You will see a significant speed up, particularly if you are using larger Sensitivity values, though we still recommend keeping Sensitivity to a low (<4) value and the Hash Value to the maximum possible value for optimal performance when searching through large amounts of NGS data.
|

