

So you just got your NGS reads back from that sequencing experiment and, wow, what a HUGE amount of data. Wouldn’t it be easier to handle if you could pare that down to just the gene/plasmid/sequence(s) you are interested in? MacVector to the rescue as it can read and filter fast/q files, even if they are compressed! Open a “reference” sequence (or create a fake composite sequence of all the sequences you are interested in, it will work just as well) then choose Database->Align to Folder. Set up the dialog something like this;

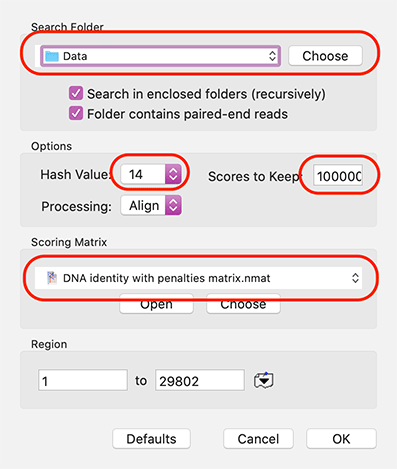
Set Search Folder: to the location of your NGS data files. Make sure Hash Value is 10 or more (for speed) and Scores to Keep is at least 1,000 or more (to make sure you don’t miss any reads) and consider the scoring matrix: for sequencing data where you are expecting essentially perfect matches, the DNA identity with penalties matrix is by far the best choice. If you are looking for reads from related organisms, other .nmat files may be more appropriate to allow for mismatches.
While the search algorithm has been optimized for use on multi-CPU machines (and for Apple M1 processors), it can still take some time to run. A 100bp sequence scanned against 3 million 133nt paired Illumina HiSeq reads takes less than 10 minutes on a typical Mac laptop, but scanning a 5 Mbp E. coli genome against 100 million reads is likely to be an overnight proposition.
When complete, you can save the matching hits by selecting all of the lines in the Folder Description List tab (Edit->Select All or command-A) then save them to a pair of matching fasta/q file by choosing Database->Retrieve to File. You will see a PAIR of files with the hits – even if only one read in the original pair matched the query file, BOTH reads are saved. This is a very powerful approach to help resolve variants and inconsistencies in NGS data. You can see from the example below that we’ve reduced a pair of 118 MB files (compressed!) to just the 2x 374 KB that are of interest to us;
